


Nucleic Acids Res | DMD治疗的新突破
- boke
- 2022-09-22
- 7:21 上午
目前治疗杜氏肌营养不良症(DMD)的方法是使用磷酰二胺吗啉寡聚物(PMO)来诱导肌营养蛋白前mRNA的外显子跳跃,使得缩短但有功能的肌营养蛋白得到翻译。由于PMO对心脏和骨骼肌的递送困难,这一策略难以应用。
为了克服这个障碍,研究人员开发了FORCETM平台,包括一个抗原结合片段,它与转铁蛋白受体1结合并与一个寡核苷酸ASO结合。研究证实,单剂量小鼠特异性FORCE-M23D聚合体可增强mdx小鼠跳跃外显子的PMO(M23D)的肌肉递送,达到剂量便可实现稳定的外显子跳跃和持久的肌营养蛋白恢复。FORCE-M23D诱导的肌营养蛋白表达在股四头肌、胫骨前部、腓肠肌、膈肌和心脏中分别达到野生型水平的51%、72%、62%、90%和77%的峰值,单次剂量为30mg/kg的PMO等效。缩短的Dystrophin定位于肉膜,表明表达的是一种功能性蛋白。
相反,单次30毫克/公斤剂量的未结合的M23D显示出较差的肌肉递送,仅能实现外显子跳跃和肌营养蛋白表达的边缘水平。重要的是,与未结合的M23D相比,FORCE-M23D治疗使肌肉得到功能性的改善。总体结果表明,FORCE聚合体是治疗DMD的一种潜在的有效方法。
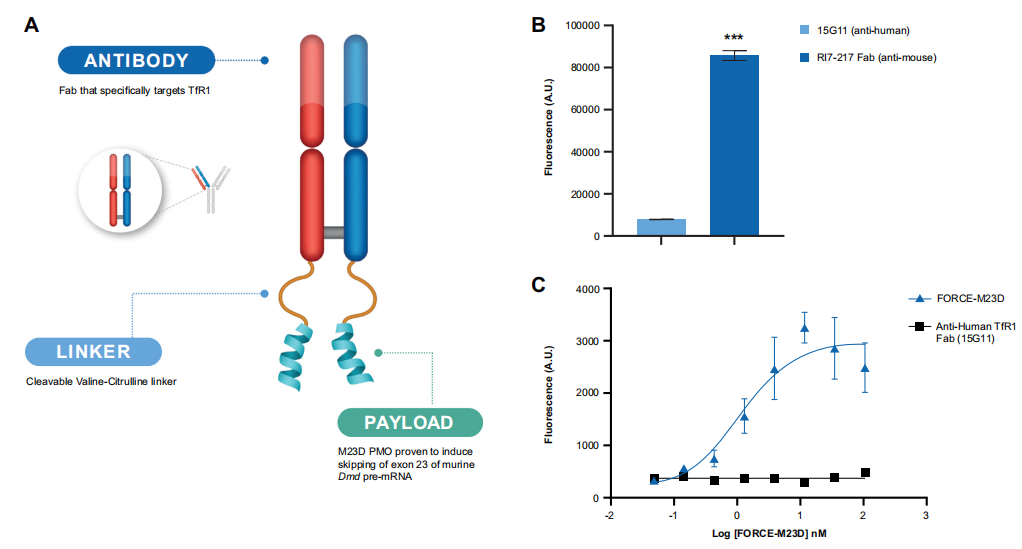
图1 FORCE-M23D结构、Fab内化动力学和FORCE-M23D在C2C12小鼠肌细胞中的TfR1结合力展示。(A) FORCE-M23D结合物的示意图。(B) 通过ELISA检测用FORCE-M23D聚合物或阴性对照的人TfR1特异性结合物与小鼠TfR1的结合。(C) C2C12肌细胞对抗鼠TfR1-Cypher5a聚合物和抗人TfR1-Cypher5e聚合物的摄取。
杜氏肌营养不良症(DMD)是一种罕见的、X连锁的、进行性的神经肌肉疾病,每3500-5000个男性新生儿中就有一个发病,导致患者在青春期前丧失行走能力,并在10~40岁之间死亡。DMD是由编码Dystrophin蛋白的DMD基因突变导致表达缺失而引起的。DMD是人类基因组中最长的基因之一(220万个碱基对),所以特别容易发生突变,其中许多突变减少或减少了肌营养蛋白的表达。
大多数(68%)的突变是组成肌营养蛋白mRNA的79个外显子中的一个或多个发生了缺失。其余的是一个以上的外显子的重复,小的indel或点突变。这些突变中的大多数中断了开放阅读框架,导致提前出现终止密码子并无法产生功能性的肌营养蛋白。肌营养蛋白通过与肌营养蛋白糖蛋白复合物相互作用,在稳定肌肉细胞的肌膜方面起着关键作用。因此,DMD患者缺乏Dystrophin会导致收缩时肌浆受损,细胞功能和信号传递失调,最终导致肌肉细胞死亡。受损的肌肉逐渐被纤维化和脂肪组织所取代,最终表现出临床症状。
外显子跳跃可以恢复DMD转录本的开放阅读框,使缩短但有功能的肌营养蛋白得到翻译。这种方法具有广泛的临床适用性,因为来自TREATNMD DMD全球数据库的数据表明,DMD患者中高达55%的各类突变和80%的肌营养蛋白基因缺失均可能适用外显子跳过疗法。一些能在DMD前mRNA中诱导外显子跳跃的反义寡核苷酸(ASO)已被批准作为DMD的治疗方案。
目前获批的治疗方法采用全身给药磷酸二酰胺吗啉寡聚物(PMO)结合ASO促使基因外显子发生跳跃,然而细胞摄取效率低下和肾脏清除速度快使其疗效大打折扣。因此,需要新的策略将促使外显子跳跃的寡核苷酸有效地输送到DMD患者的肌肉中。
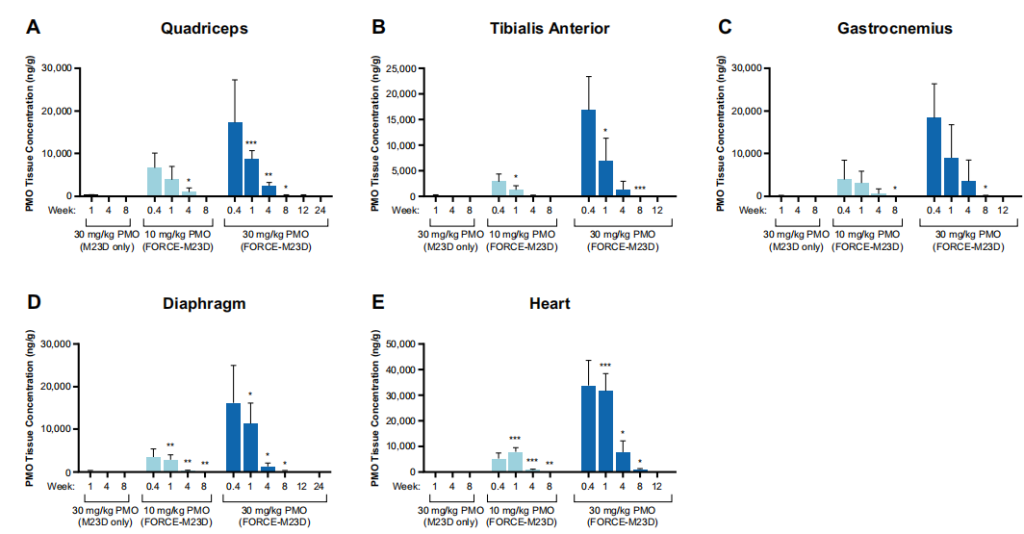
图2 单剂量的M23D,但不是未结合的M23D,在mdx小鼠的骨骼肌和心肌中诱导出剂量依赖的PMO释放。
FORCE™平台的开发是为了克服向肌肉输送寡核苷酸的限制。它利用了转铁蛋白受体1(TfR1)的特性,该受体在骨骼肌、平滑肌和心肌细胞表面表达,对这些组织的铁吸收至关重要。TfR1通过介导铁结合的转铁蛋白的内吞作用来控制细胞的铁平衡,并以17分钟的循环速度返回到细胞表面。
虽然针对TfR1的二价抗体改变了向细胞表面的循环,但抗原结合片段(Fab)不影响受体表面的表达、内化或降解,使其成为TfR1靶向的首选方法。FORCE平台核心的TfR1靶向Fabs是用缬氨酸连接剂与ASO连接的,它在血浆中是稳定的,并在内涵体中被酶解,使寡核苷酸有效载荷在细胞膜中释放,并随后靶向结合对应基因。
本研究利用FORCE平台将PMO传递到成熟的DMD模型mdx小鼠的肌肉中。FORCE-M23D(图1A)是一种小鼠特异性的Fab-PMO结合物,由Fab片段组成,对小鼠转铁蛋白受体1具有特异性,并且已知不会干扰转铁蛋白的结合,与PMO有效载荷(M23D)结合,旨在跳过第23外显子。
研究数据显示,单剂量的FORCE-M23D可实现强大而持久的外显子跳过,恢复骨骼和心肌中功能正常的肌营养蛋白,并改善mdx小鼠的运动功能,而单剂量的未结合的M23D则没有任何益处。
美国已批准基于PMO诱导的外显子跳转疗法,用于治疗具有可跳过外显子51、53和45的突变的DMD患者。虽然外显子跳转方法在治疗DMD方面具有很大的前景,但已批准的疗法显示,收缩蛋白恢复的增加非常有限,如果有的话,功能性改善也不大。
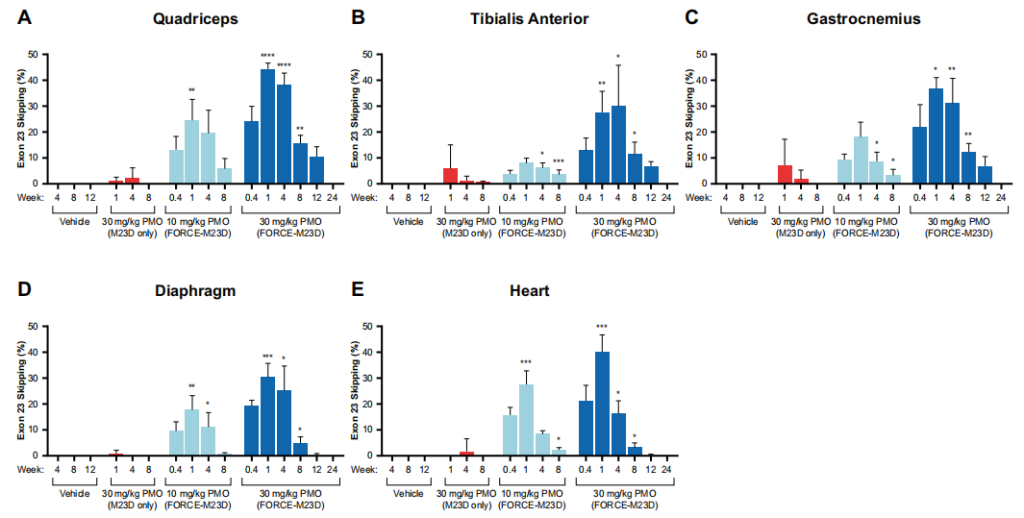
图3 单一剂量的FORCE-M23D,而不是未结合的M23D,在mdx小鼠的骨骼和心脏肌肉中诱导出剂量依赖的Dmd基因外显子23跳跃。
实现对肌肉的有效递送仍然是开发临床有效疗法的一个瓶颈。ASO的递送通过化学修饰、使用替代的化学骨架和与递送分子(如肽)结合而得到改善。PMO结构中吗啉环取代了脱氧核糖基,磷酸二酯连接取代了磷酸二酯基内连接,与原生DNA或RNA相比,对核酸酶和蛋白酶活性更稳定。然而,大量已发表的研究表明,由于心肌和骨骼肌的吸收能力差,PMO作为DMD的治疗方法存在缺陷。因此,单次PMO剂量的作用时间很短,治疗需要高的药物剂量或者不断给药,以达到治疗效果,但仍受到不一致的肌营养蛋白恢复的阻碍。
本研究开发了一种TfR1靶向的FORCE-M23D聚合物在体外以纳摩尔的亲和力与小鼠TfR1结合,并导致TfR1介导的吸收进入C2C12小鼠肌细胞。结果显示FORCE-M23D向mdx小鼠全身给药时,能够诱导Dmd基因第23号外显子跳跃的PMO进入骨骼和心肌。PMO的组织浓度在单次用药后很快达到了一个峰值,随后外显子23跳跃达到峰值。虽然组织暴露和外显子23跳跃水平在达到峰值后不久就下降了,但在用药后4周和8周,在心肌和骨骼肌(包括横膈膜)观察到高水平的肌营养蛋白恢复。这与肌营养蛋白在骨骼肌中相对较长的半衰期一致。给予FORCE-M23D比同等剂量的未结合的M23D有明显更多的暴露和更大的药理作用。
虽然两种治疗方法之间的比较并非都达到了统计学意义,但这可能是由于mdx小鼠的个体差异性造成的。此外,非结合型M23D导致心肌和骨骼肌的不一致、边缘暴露和分子以及功能药理反应,这与非结合型PMO给药的已知缺陷相同。
对股四头肌切片的免疫荧光分析补充了对蛋白质表达的观察,发现肌营养蛋白定位在肉膜上,即其生理作用部位,表明缩短的肌营养蛋白在功能上是健全的。重要的是,在膈肌和心脏切片中,大量的肌营养蛋白定位在细胞膜上,尽管观察结果是定性的。与股四头肌不同,横膈膜和心脏中的肌营养蛋白恢复总量似乎在用药后8周迅速减少。这一观察可能是由于横膈膜和心脏持续收缩的机械压力造成的。总之,这些发现表明,FORCE-M23D向心脏和骨骼肌提供了大量的PMO,而且从跳跃的转录本翻译的肌营养蛋白可能具有功能。
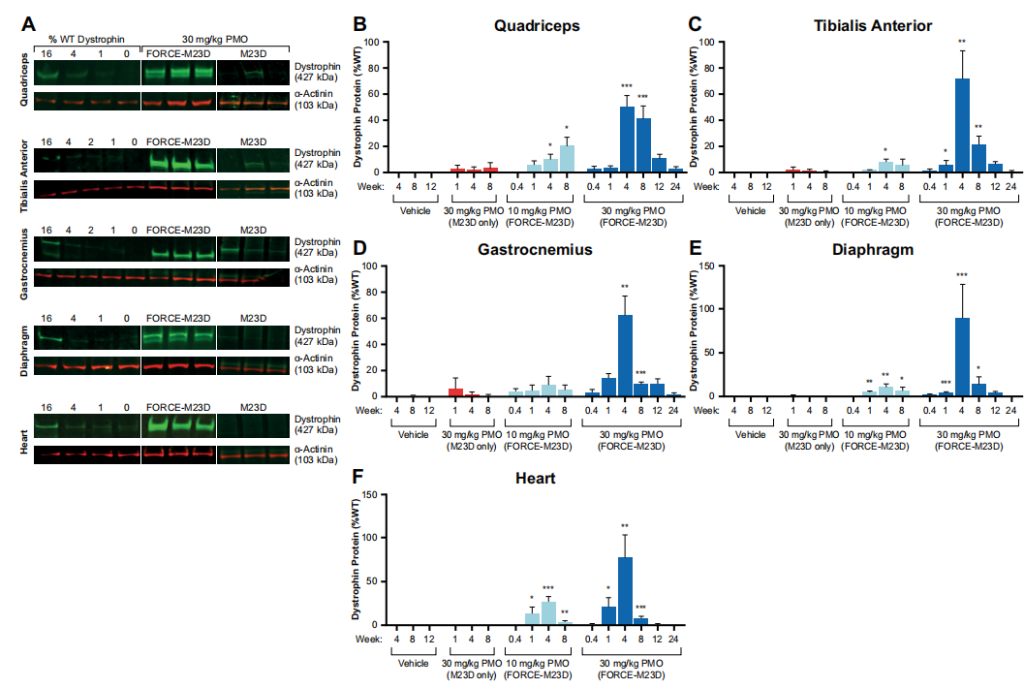
图4 单一剂量的FORCE-M23D,而不是未结合的M23D,诱导mdx小鼠骨骼和心肌中剂量依赖性的dystrophin蛋白的表达。
给予单剂量的FORCE-M23D后,观察到的药效学反应的持续时间和肌营养蛋白在肌节的定位程度与膜完整性的改善相关。此外,血清CK水平的降低与功能型mdx表型的纠正相结合,表明FORCE-M23D治疗恢复了有意义的功能型dystrophin蛋白的数量。
FORCE-M23D导致了卓越的分子和表型纠正,而非结合的M23D治疗取得了适度的效果,证实了FORCE平台加强了寡核苷酸治疗剂对骨骼肌和心肌递送的效果。基于这些数据推测,基于FORCE平台的疗法可能使DMD患者得到更大的临床获益。
目前治疗DMD的药物eteplirsen(EXONDYS51,Sarepta Treateutics,Cambridge,MA)是一种30核苷酸的PMO,可促使DMD转录本第51外显子的跳跃。虽然eteplirsen已被批准在美国使用,但验证和描述其预期临床益处所需的上市后验证性试验尚未完成。全身给药导致靶组织的非特异性摄取,在动物模型中的工作很好地证实了这种方法不能恢复心脏中肌营养不良蛋白的表达。
这一点至关重要,因为心血管并发症是DMD患者发病率和死亡率的主要原因。考虑到FORCE-M23D诱导的mdx小鼠心肌肌萎缩蛋白恢复的幅度,FORCE平台具有向心脏稳定输送PMO的潜力,能够恢复DMD患者心脏中Dstrophin蛋白的表达,并有可能降低继心脏不良事件的死亡率。
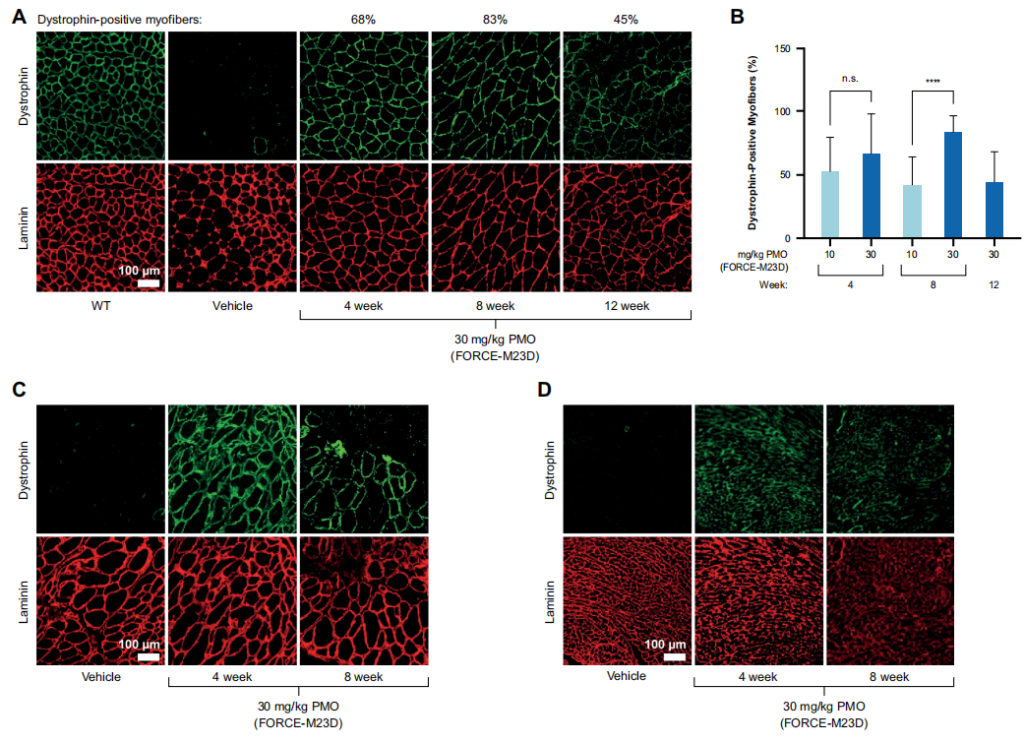
图5 单一剂量的FORCE-M23D足以恢复mdx小鼠骨骼和心脏肌肉中肌营养蛋白的表达。
为了解决目前外显子跳跃PMO分子的局限性,提高其作为治疗DMD的潜力,人们开发了细胞穿透肽的偶联策略(CPP-PMO)。多项体外和体内研究的结果表明,与未结合的PMO相比,CPP-PMOS显示出更好的细胞内化、全身给药效率、持续的抗肌营养不良蛋白的产生,以及更高的效力。
有研究发现mdx小鼠在最后一次给药后2周,以每周一次25 mg/kg的CPP-PMOS产生外显子跳跃,或以75mg/kg的单次给药,在骨骼肌中显示出有效的外显子跳跃和dystrophin表达,但在心脏中没有表达。在另一项研究中,Betts等人使用PMO内在化多肽(Pip6-PMO)并报道了mdx小鼠心脏中的外显子跳跃和dystrophin的表达。因为在单次注射10 mg/kg后可以观察到适度的dystrophin表达,所以实施了一个总计9个剂量的较长的治疗方案,导致隔膜(77%)和TA(86%)内高水平的外显子23跳跃。
然而,药物对心脏的影响并不大,只有32%的人出现外显子跳跃,28%的人恢复了dystrophin的表达。因此,尽管多肽结合改善了PMOS的全身递送,但对心肌的穿透仍然是一个挑战。与CPP-PMO不同,FORCE-M23D不包含破坏膜稳定性的结构,依靠连接子的切割从结合物中释放M23D有效载荷,然后PMO从内体自发逃逸。
利用这一策略,在mdx小鼠体内注射等量30 mg/kg的pmo-M23D后,在给药4周后,心脏中40%的外显子跳跃和77%的dystrophin蛋白表达达到峰值。这些观察结果表明,与CPP-PMO和其他利用细胞穿透策略进行有效载荷摄取的方式相比,TfR1介导的摄入是一种更好的心肌组织递送方式。
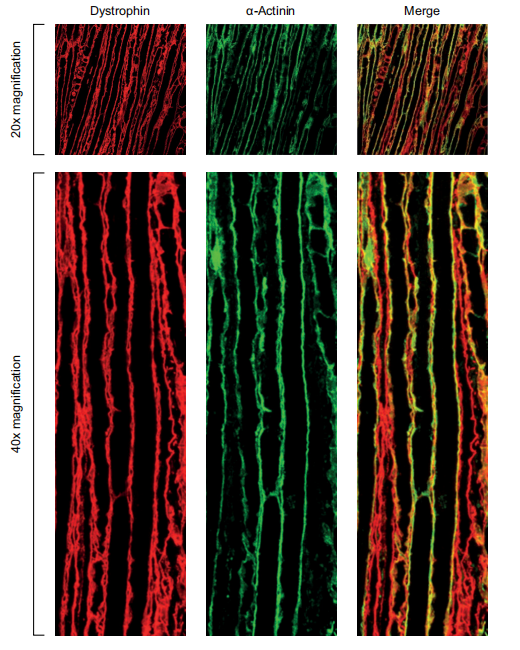
图6 由FORCE-M23D恢复的Dystrophin定位于mdx小鼠的肌肉膜上。
虽然CPP-PMO结合可以增加对肌肉的转运,但多肽部分在肝脏和肾脏等器官中具有已知的安全风险,可能导致毒副作用,可能是由多肽的阳离子性质驱动的。许多其他因素,如治疗时间、剂量、全身给药频率,以及用于测试CPP-PMOS的动物模型,也可能导致副作用出现。因此,使用较低剂量或允许两次给药间隔较长可能有助于降低其中一些风险。
目前,有一项考察CPP-PMO SRP-5051用于治疗可跳过外显子51的DMD患者的情况的临床试验正在进行,这可能会为CPP-PMO模式提供临床相关的安全性和药理学指导(临床试验.gov,NCT04004065)。值得注意的是,单剂量的FORCE-M23D在mdx小鼠身上实现了与PPMOs研究中观察到的类似水平的跳跃,在骨骼肌和心肌中的药理效应长达12周。作用时间的延长和较低的有效剂量水平支持这样一种观点,即FORCE平台可能具有比依赖细胞穿透肽的递送机制更优秀的治疗指数(半数致死量(LD50)与半数有效量(ED50)的比值)。
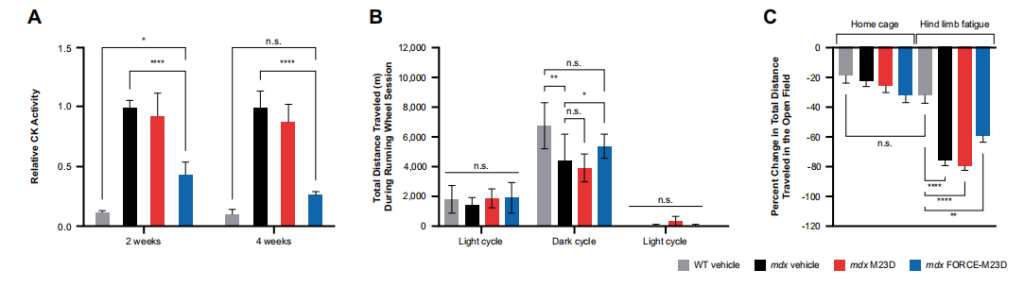
图7 用FORCE-M23D治疗,而不是用未结合的M23D,会促使mdx小鼠的肌肉功能改善。
mdx小鼠是DMD常用的动物模型;然而,由于其营养不良表型没有DMD患者的表现那么严重,它可能不是代表人类疾病的最佳模型,这是当前研究的局限性。然而,目前的数据表明,与利用非结合寡核苷酸的方法相比,单剂抗TfR1 FORCE-M23D结合物可以提供更好的外显子跳跃和dystrophin表达,以及更大程度的肌肉功能改善。DMD是一种慢性进展性疾病,需要外显子跳跃PMO的持续治疗。虽然单剂量的FORCE-M23D显示出比非结合有效载荷更好的药效学特征,但更多的重复剂量研究将是必要的,以了解FORCE提供持久的肌营养不良蛋白恢复和功能益处的能力。
总之,FORCE平台结合了生物药物的药理学特性和寡核苷酸疗法的特点,有可能为DMD患者提供一种有效、可重复使用并且可定量的治疗方案。


