


Nature Reviews | 人类基因组结构变异(SVs)的多态性及其影响
- boke
- 2025-10-14
- 4:42 下午
该文章总结了人类群体中生殖细胞结构变异的现有研究进展。
第一,探讨结构变异的突变特性,包括其多样性、复杂性和突变率。
第二,描述结构变异在全球人类群体中的分布模式,重点关注自然选择的作用。
第三,总结结构变异导致全基因组编码区和非编码区功能改变的方式。
第四,综述结构变异在罕见病、常见病及复杂疾病中的作用,并展望技术发展在该研究领域的应用前景。
最后,列举了未来几年有望成为研究热点的三大结构变异研究前沿方向。
一
核心背景与定义
根据变异涉及的 DNA 核苷酸数量,人类遗传变异通常分为三类:单核苷酸变异(single-nucleotide variants, SNVs)、短插入 / 缺失变异(indels,1-50 bp)和结构变异(structural variants, SVs,≥50 bp)。
SVs在人类群体中普遍存在,其突变机制具有异质性;包括拷贝数变异(CNVs,缺失 / 重复)、插入(移动元件插入(如长散在核元件 1(LINE1)和短散在核元件(Alu))、新的非参考序列插入(如病毒 DNA 插入)、线粒体 DNA 的核插入,以及串联重复序列扩张(例如,短串联重复序列(STRs,重复单元为 1-10 bp)和可变数目串联重复序列(重复单元为 10-100 bp))、倒位、易位(导致基因组重排但不改变拷贝数的结构变异)及复杂结构变异(如染色体重生),是人类基因组核苷酸多样性的主要来源(平均每基因组改变 32.1 Mb,远超短变异的 6.7 Mb)。
SVs 是进化适应的重要基础,同时也是疾病的关键驱动因素(如 21 三体与唐氏综合征、22q11.2 缺失与迪乔治综合征)。对大型人群的深度(≥30倍覆盖度)WGS表明,人类群体中可能有数千万个独特的SVs,其中大多数仅出现在全球一个或少数个体中。
尽管复杂结构变异整体具有异质性,但它们存在三个共同趋势:
第一,倒位是复杂结构变异的常见特征,且这些复杂倒位的一端或两端常伴随拷贝数变异,提示倒位可能是介导复杂结构变异形成的因素之一 ;
第二,复杂结构变异在重复序列中富集,表明涉及重复序列的重排机制可能在新复杂结构变异形成中具有特殊作用 ;
第三,重排复杂性差异显著——尽管目前人类生殖细胞中已鉴定的复杂结构变异几乎均相对简单(仅涉及 2-3 个片段或断裂点),但也有数十个极端复杂的生殖细胞结构变异被报道,这类变异统称为 “染色体重生”(chromoanagenesis)。

Fig. 1 | Human SVs span a broad mutational spectrum.
二
结构变异的关键特性
突变机制与类型
1.核心机制:
非同源末端连接(non-homologous end-joining, NHEJ)、复制叉停滞 – 模板切换、微同源性介导的断裂诱导复制、非等位同源重组(non-allelic homologous recombination, NAHR),解释了目前人类基因组中已发现的多数CNVs;可移动元件插入由转座 / 逆转座引起,串联重复扩张由 DNA 聚合酶滑移导致。
最简单的结构变异断裂点为无同源性的平末端,通常由 DNA 双链断裂后通过NHEJ修复形成 。NHEJ事件的断裂点常存在 “痕迹”,表现为小片段(<10 bp)缺失或非模板插入,这是由断裂修复不完全导致的 。
另一种结构变异断裂点在断裂末端存在短片段(<70 bp)序列同源性(即 “微同源性”),这类变异常通过复制叉停滞 – 模板切换或微同源性介导的断裂诱导复制形成 。
较长的同源序列对(通常长达数百或数千核苷酸)可通过NAHR形成拷贝数变异、倒位或复杂结构变异,具体类型取决于同源序列的方向。
可移动元件插入由内源性可移动元件的转座或逆转座导致 ,而串联重复序列扩张则由复制过程中 DNA 聚合酶滑移引起 。
复杂结构变异的形成机制更为多样,包括涉及倒位 DNA 重复的多步突变级联反应,以及细胞分裂过程中染色单体错误分离进入微核等。
此外,某些结构变异形成机制可能仅存在于高度重复且高突变率的基因组区域,如着丝粒附近或近端着丝粒染色体臂上的大片段(>100 kb)微卫星序列和 DNA 重复阵列 ,这些机制目前尚未明确,但有望通过泛基因组组装在未来得到阐明 。
2.类型多样性:
典型 SVs:CNVs(双等位 / 多等位)、插入、倒位(平衡性,对染色体结构影响显著);
复杂 SVs:少数人类结构变异存在显著的重排复杂性—,通常表现为在同一突变事件中,拷贝数变异(CNVs)或倒位与一个及以上其他断点同时出现,被定义为 “复杂结构变异(complex SVs),如染色体重生(涉及数十个断裂点,罕见但致病性强)。
群体遗传学特征
1.分布模式:每个人类基因组携带约 2×10⁴个 SV,其中 > 90% 为插入 / CNVs;多数 SV 低频(等位基因频率 <1%),但个体中> 95% 为常见 SV(≥1%)。
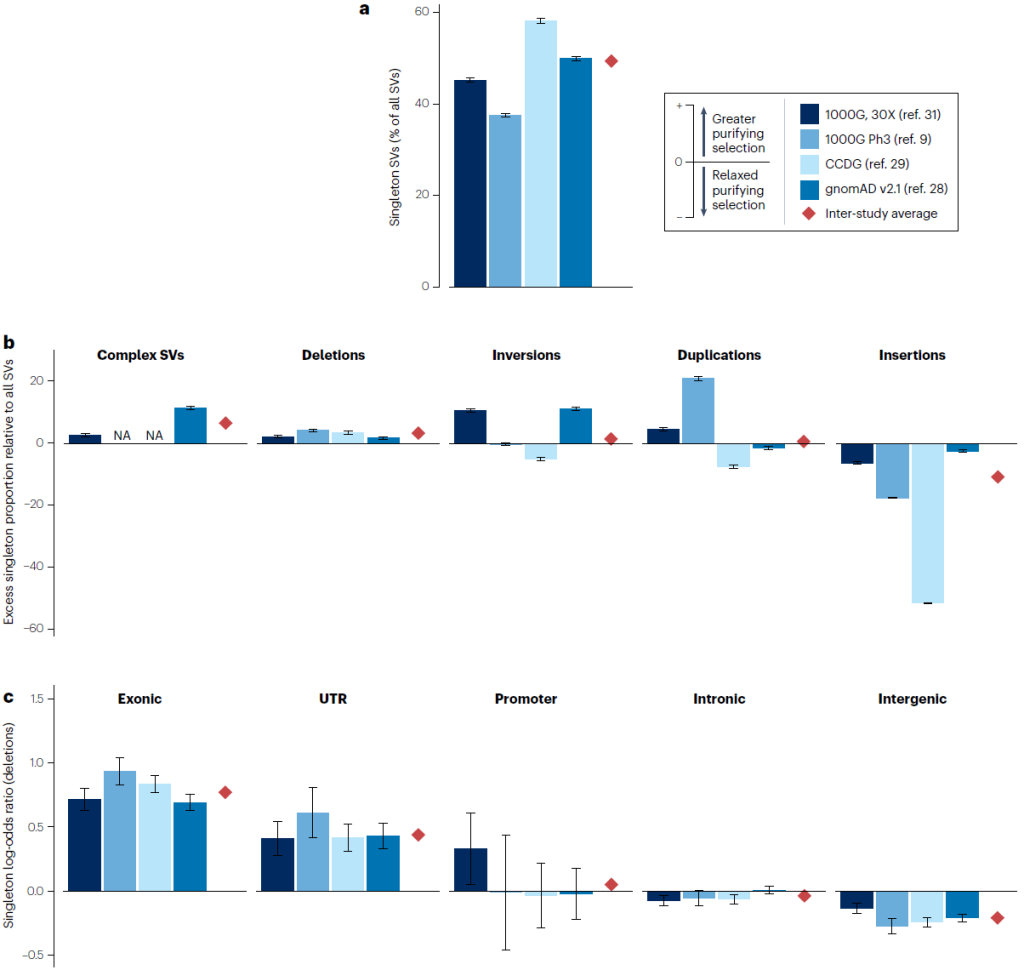
Fig. 2 | Most SVs experience purifying selection in the general population.
2.自然选择:
负选择:SV 大小与选择强度正相关(大片段 SV 频率更低),缺失比重复面临更强负选择,破坏基因的 SV 比非破坏基因的 SV 选择压力更强;
正选择:少数 SV 因适应优势被选择(如 AMY1 拷贝数增加适应高淀粉饮食,HP 外显子缺失降低胆固醇)。
三
检测结构变异的技术与算法
相比于参考基因组,人类平均每个基因组中存在数千个结构变异,但每个基因组中识别出的结构变异具体数量与类型,取决于用于检测结构变异的技术与算法。
短读长研究报告,人类平均每个基因组存在 9000-13000 个结构变异。相比之下,长读长全基因组测序研究对每个基因组中结构变异的检测灵敏度更高,结果显示平均每个基因组存在22000-26000个结构变异。插入型变异与拷贝数变异(CNVs)在每个基因组的所有结构变异中占比均超过 90%。此外,人类平均每个基因组还存在数百个平衡性结构变异与复杂结构变异。 任何个体基因组中存在的>95%的SVs是常见的(等位基因频率≥1%)多态性SVs。
四
功能影响与疾病关联
功能作用方式
编码区影响:直接破坏基因(导致 LoF 或拷贝数增加)、产生基因融合、改变 mRNA 剪接(如可移动元件插入); 非编码区影响:破坏顺式调控元件(CREs)、改变三维基因组结构(如破坏拓扑关联结构域 TADs),且 SV 的长距离调控效应(影响≥250 kb 外基因)比短变异更显著。
最新的 GTEx 结构变异研究发现,1271 个常见结构变异(其中 72% 为非编码区变异)与邻近基因的 RNA 表达水平显著相关,这类变异被称为表达数量性状位点(eQTLs)。
基因对 “功能丧失型变异” 的耐受程度呈连续分布,而 “剂量敏感性” 作为一种特殊的突变耐受限制,对拷贝数变异(CNVs)及其他非平衡性结构变异尤为重要。
剂量敏感性描述的是基因或位点的 “拷贝数” 与 “适合度” 之间的关系,既包括由功能丧失型结构变异导致的 “单倍体不足”(haploinsufficiency,即对拷贝数减少的不耐受),也包括由拷贝数增加型结构变异导致的“三倍体敏感性”(triplosensitivity,即对拷贝数增加的不耐受)。目前仅 3 个基因(APP、LMNB1 和 PLP1)被证实具有三倍体敏感性,长读长WGS在人群规模的应用对于理解嵌入在高度重复基因组位点中的数百个蛋白编码基因的剂量敏感性至关重要。
疾病贡献
1.罕见病 / 孟德尔疾病:
基因组疾病:由复发性 CNVs 引起(如 15q11-q13 缺失与安格尔曼综合征),单次 SV 可影响多个基因;
孟德尔疾病:SV 直接破坏疾病基因(如 L1 插入导致血友病 A)或通过位置效应(如非编码缺失破坏 LncRNA Maenli 导致肢体畸形)。
2.常见复杂疾病:与 GWAS 位点连锁不平衡的 SV 可能为部分因果变异(如 C4 位点 SV 与精神分裂症 / 自身免疫病相关),但贡献相对温和(仅 3.2%-14.2% GWAS 位点由 SV 解释)。
3.临床诊断:CNVs 是发育障碍 / 智力障碍的主要病因(诊断率 26%-29%),微阵列是一线检测手段;全基因组测序可提升诊断率(如英国 NHS 研究中 9.8% 诊断来自 SV)。
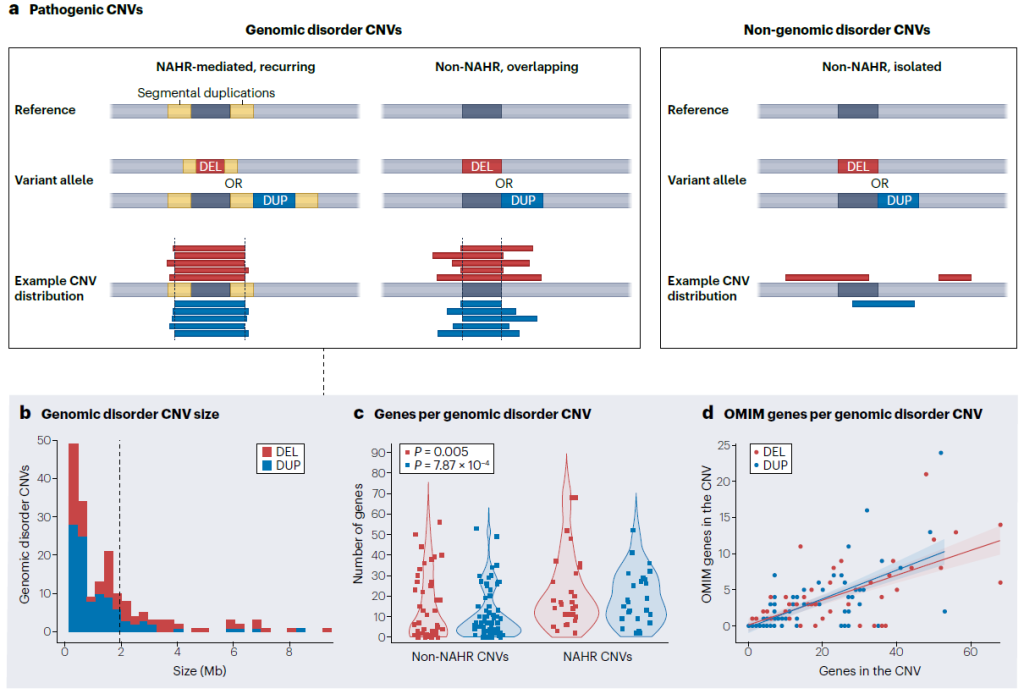
Fig. 3 | Properties of large pathogenic CNVs associated with genomic disorders.
五
未来研究前沿
复杂重复位点 SV 的解析:利用长读长和泛基因组图谱,揭示 10% 高重复区域中隐匿性 SV 的功能与疾病关联。
一方面,借助长读长测序与基因组组装算法,构建 “泛基因组图谱”,精准呈现复杂位点的群体水平结构多样性;另一方面,需将传统基于线性参考基因组的注释体系(如基因、转录本定义及染色质数据集)适配到泛基因组图谱中,为后续功能解析奠定基础。同时,要通过大规模人群长读长测序与临床数据结合,填补重复区域结构变异的注释空白,明确其与疾病的关联。
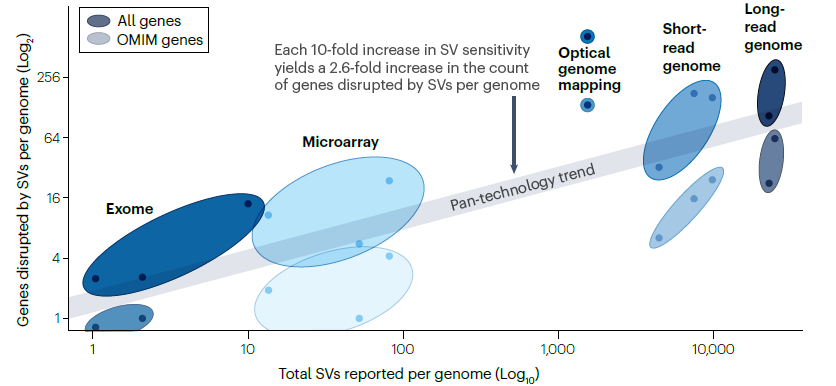
Fig. 4 | Yield from SV analyses across genomic technologies.
SV 突变率与剂量敏感性图谱:需 10 万 – 100 万样本构建精准模型,区分单倍体不足(LoF 敏感)与三倍体敏感性(拷贝数增加敏感)基因。
第一,整合海量基因组数据集与分布式计算技术,训练统计或机器学习模型,明确基因组特征与结构变异发生的关联,确定结构变异高频发生的基因组语境;第二,由于人类生殖细胞新生结构变异发生率低(每代不足 1 个),需建立 “中性结构变异筛选标准”(类似同义短变异),以完善突变率模型参数;最后,需扩大样本量以构建全面的剂量敏感性图谱,例如基于现有基因组聚合数据库(gnomAD)数据估算,需至少 10 万个样本才能使结构变异推导的单倍体不足 metrics 精度媲美短变异。
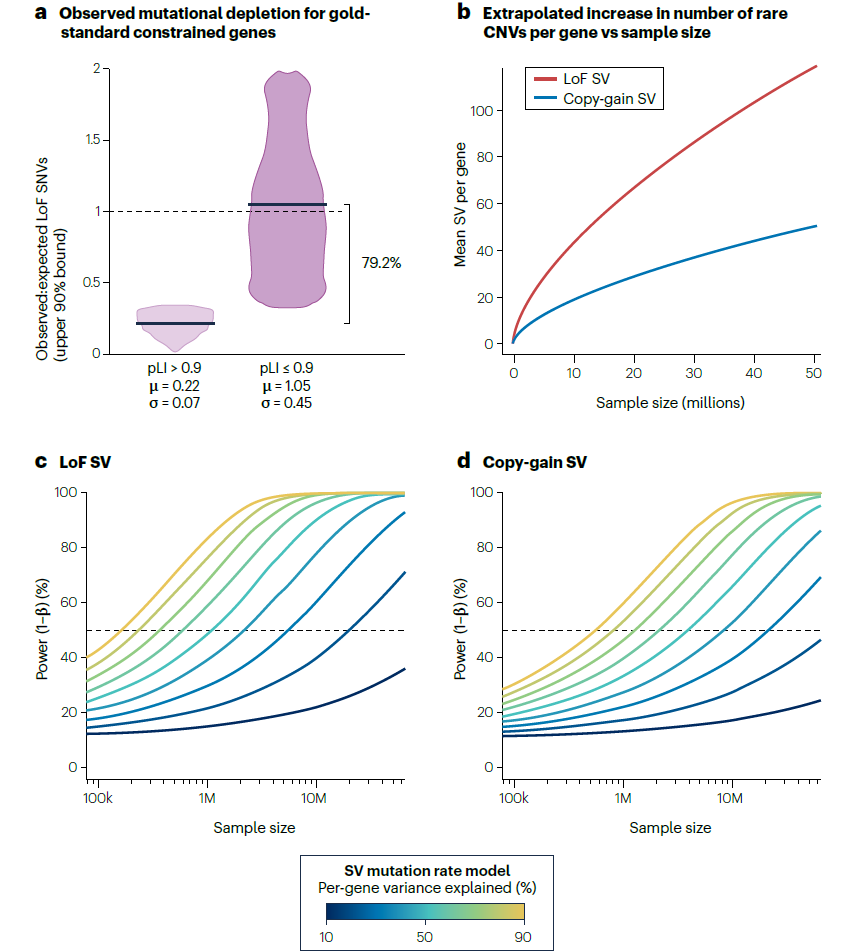
Fig. 5 | Projections for dosage sensitivity mapping in the human genome.
SV 与短变异的整合应用:
在技术层面,可利用结构变异与短变异的连锁不平衡关系,将长读长测序发现的结构变异通过基因型填充技术应用于大规模短读长全基因组关联研究数据集中,提升常见结构变异的疾病关联分析效率。 分析层面,开发统计方法联合分析 SV 与短变异的疾病效应,建立泛基因组背景下的临床解读标准,推动基因组测序在诊断中的应用。
伯科生物长读长靶向捕获平台
——遗传病结构变异

已知的DMD基因致病变异中,基因片段重复或缺失导致占比65-80%,点突变占比20%-35%,此外倒位等复杂结构变异也可能导致疾病,因此对结构变异的精确检测尤其重要。利用伯科DMD基因三代捕获测序Gene Panel,郑州大学第一附属医院与希望组研究人员报道了两例均为DMD基因56-61 exons重复,却拥有截然不同的变异特征和表型的案例。
——肿瘤结构变异

伯科生物助力北京大学人民医院和北京希望组等机构的研究者们开发了28-Gene Panel,并利用Oxford Nanopore Technologies和Pacific Biosciences长读长测序平台,对19名乳腺癌患者的肿瘤、癌旁组织和血液样本进行长读长基因组和转录组测序,分析了乳腺癌相关28个基因的结构变异。
结果表明,一些体细胞的结构变异在选定的基因中反复出现,并且大多数发生在非外显子区域。研究发现不仅有单核苷酸相关的热点区域,也有结构变异反复出现的热点区域。本研究通过采用靶向长读长测序来识别乳腺癌患者的SV,并证明了该方法在临床应用中有巨大潜力。
参考文献:
1.Ryan L. Collins, Michael E. Talkowski. Diversity and consequences of structural variation in the human genome. Nature Reviews Genetics 26, pages443–462 (2025). https://doi.org/10.1038/s41576-024-00808-9
2. Ling C, Dai Y, Geng C, Pan S, Quan W, Ding Q, Yang X, Shen D, Tao Q, Li J, Li J, Wang Y, Jiang S, Wang Y, Chen L, Cui L, Wang D. Uncovering the true features of dystrophin gene rearrangement and improving the molecular diagnosis of Duchenne and Becker muscular dystrophies. iScience. 2023 Oct 30;26(12):108365.
3. Hu T , Li J , Long M , et al. Detection of structural variations and fusion genes in breast cancer samples using third-generation sequencing. 2021.