


Nature Genetics 丨超18万例2型糖尿病的跨种族遗传研究 人群多样性的发现和转化的重要性
- boke
- 2022-08-18
- 7:21 上午
全球2型糖尿病(T2D)患病率在过去30年翻了两番,2015年影响了约3.92亿人。尽管具有世界范围的影响,但绝大多数的T2D GWAS主要以欧洲血统人群为特征。对欧洲以外人群疾病风险变异研究的缺乏,限制了对疾病生物学的全面了解和治疗学的发展。实施个性化的疾病管理方法取决于不考虑祖先因素对个体风险的准确预测。
2022年5月12日Andrew P. Morris团队在《Nature Genetics》上发表了题目为“Multi-ancestry genetic study of type 2 diabetes highlights the power of diverse populations for discovery and translation”的研究,通过跨种族GWAS荟萃分析,增强了在不同人群中的T2D预测的能力。
该研究人员通过180,834例T2D患者和1,159,055例对照(48.9%非欧洲后裔),对糖尿病GWAS祖先多样性进行整合。跨种族GWAS荟萃分析鉴定了237个全基因组显著性位点(P<5×10–9),划分为338个不同的关联信号。随着样本量的增加和群体多样性的扩大,精细定位信号增强,54.4%的T2D关联信号定位到单一突变。这种改进的精细定位能够系统地评估候选基因和T2D关联信号介导的分子机制,为功能研究奠定了基础。跨种族遗传风险评分增强了在不同人群中的T2D预测的通用性。该研究推动了T2D GWAS更有效的临床转化、为改善全球健康迈出了重要一步。
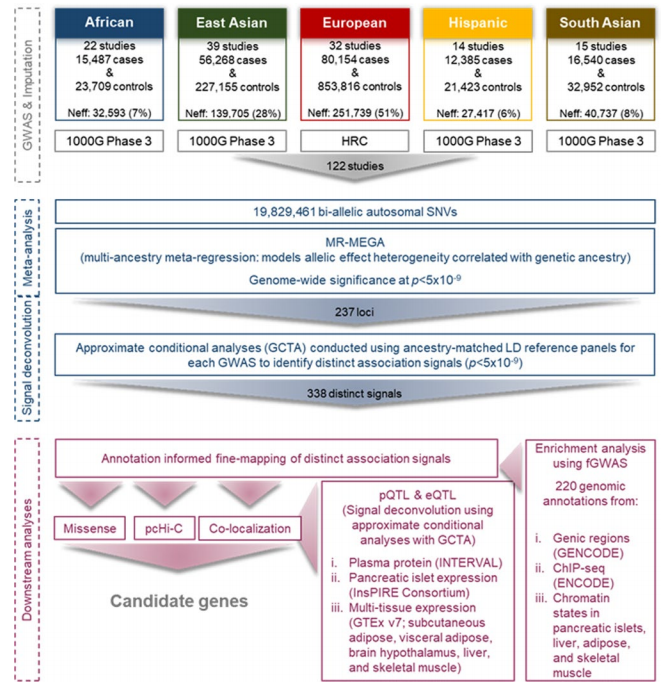
图1 研究概述
结果
通过MR-MEGA的跨种族荟萃分析,模拟了祖先遗传相关的等位基因异质性效应。研究人员识别了277个与T2D相关的位点(P<5×10−8)。在277个位点中,有11个在最近发表的T2D GWAS分析中没有报道,占跨种族荟萃分析总有效样本量的78.6%。在东亚和欧洲的祖先特异性联合分析中,分别有100个和193个位点具有全基因组显著性,其中94个(94.0%)和164个(85.0%)lead SNVs在跨种族荟萃分析中显示了更强的相关性(更小的P值)。
通过使用来自1000 Genomes Project作为参考的单倍型估算五个祖先类群中独立SNVs的有效数量,得出了跨种族全基因组显著性阈值为P<5×10−9。跨种族荟萃分析中报告的277个位点中,有237个达到了更严格的显著性阈值并用于下游分析。使用祖先群体匹配的连锁不平衡(LD)作为参考对每个GWAS进行近似条件分析,将237个位点划分为338个不同的信号,每个信号用一个SNV指数表示。研究证明,在混合的祖先群体中进行近似条件分析对不同关联信号的等位效应估计参考的选择是可靠的。
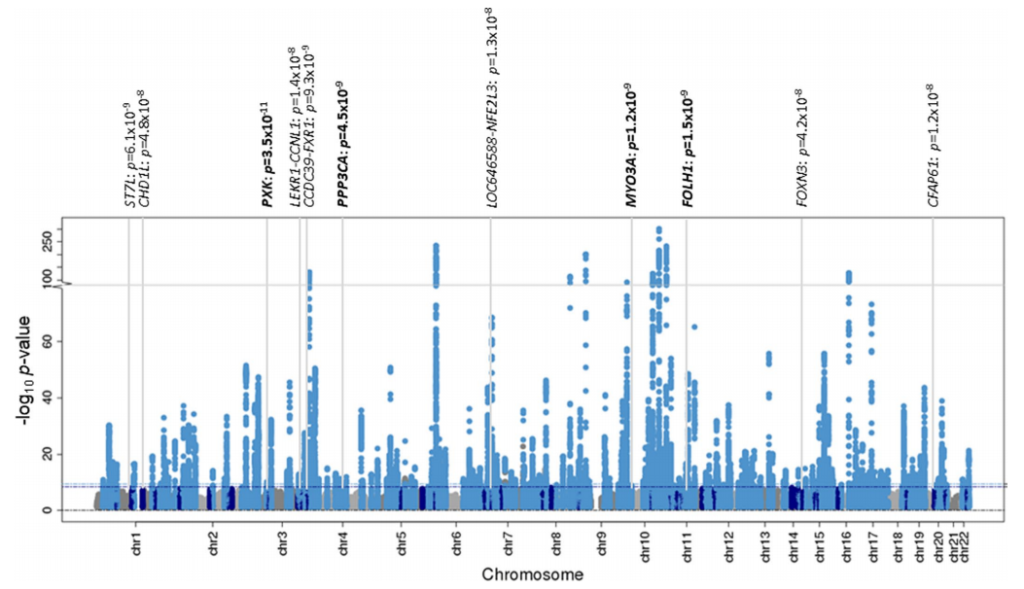
图2 跨种族荟萃分析(MR-MEGA)得到全基因组T2D关联的曼哈顿图
研究发现跨种族荟萃分析99%可信集(266个(78.7%)不同T2D关联信号)比欧洲祖先特异性关联分析中要小,而26个(7.7%)信号在两个分析中都被解析为单个SNV。与欧洲和东亚血统相比,跨种族的荟萃分析中因果变异定位也更精确,突出了代表人数不足的的祖先群体(非洲、西班牙和南亚)对精细映射分辨率的重要贡献。
相对于欧洲祖先特异性荟萃分析(119个信号(44.7%)),在降采样的跨种族荟萃分析中(137个信号(51.5%))得到了更好相关信号。这些结果强调了在跨种族荟萃分析中,不同人群对因果变异定位的价值,强调了样本量增加、祖先群体间LD结构和等位基因频率分布的重要性。
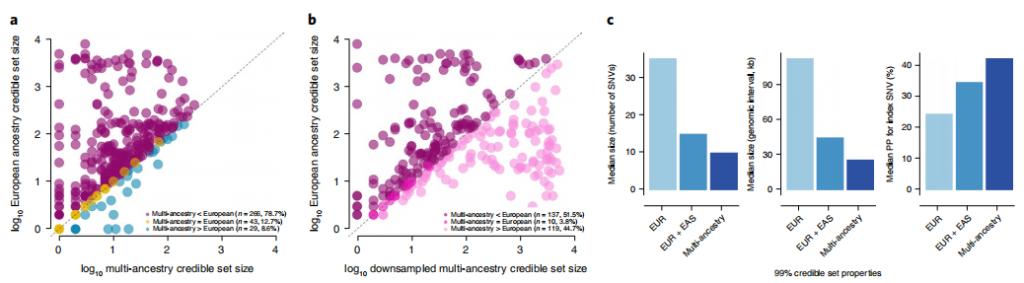
图3 祖先特异性和跨种族荟萃分析不同关联信号的精细定位分辨率比较
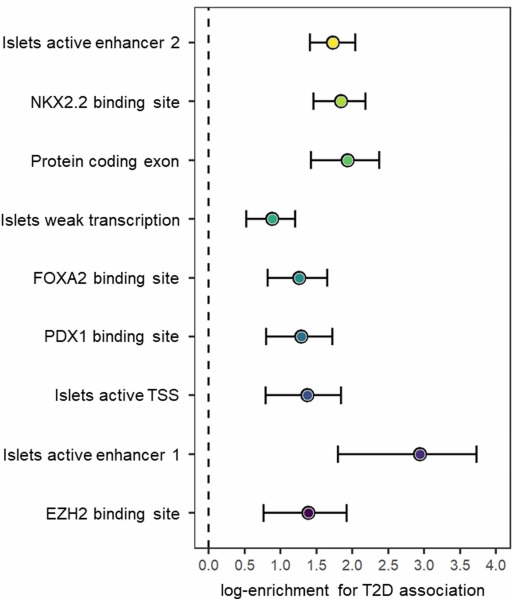
图4 不同T2D关联信号中功能和调控注释汇总统计
通过注释信息,184个SNVs中有19个为T2D相关性错义变异,后验概率>50%。其中,MYO5C位点的MYO5C p. Glu1075Lys (rs3825801, P=3.8×10-11,π=69.2%)和CYTIP位点的ACVR1C p. Ile482Val (rs7594480, P=4.0×10-12,π=95.2%)可能是新的候选致病基因。ACVR1C编码转化生长因子β受体ALK7,过表达可诱导胰腺β细胞生长抑制和凋亡;ACVR1C p.Ile482Val先前被认为与体脂分布有关。跨种族荟萃分析通过在不同种群间进行精细映射与先前报道的信号得到更好关联,包括SLC16A11、KCNJ11-ABCC8和ZFAND3-KCNK16-GLP1R。
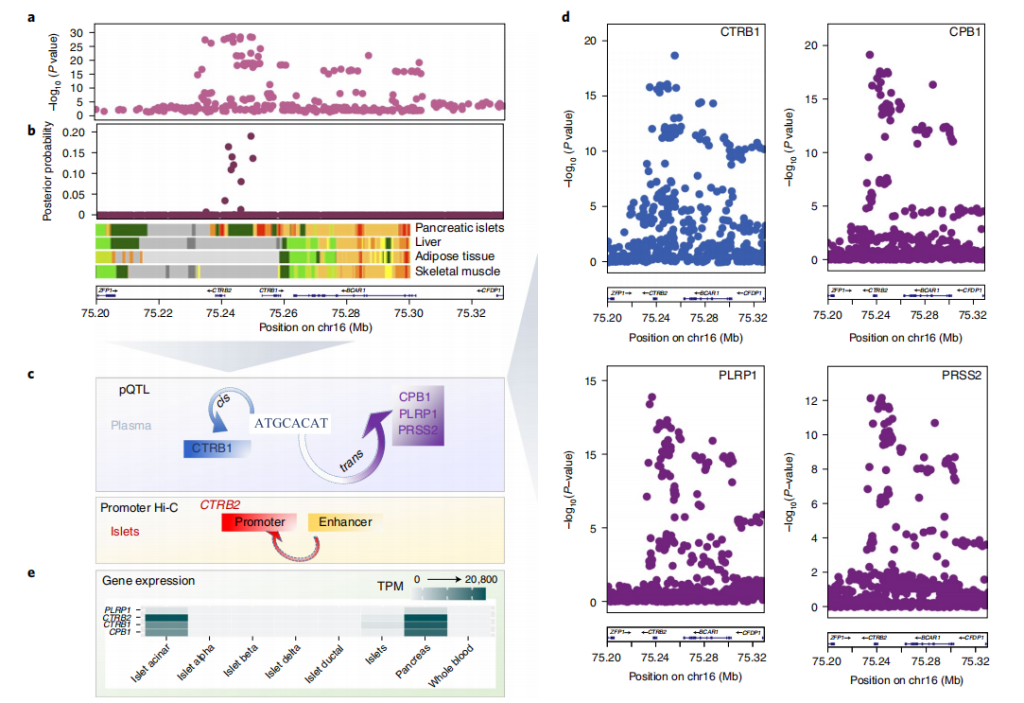
图5 BCAR1位点的T2D相关信号与多个血液循环pQTL共定位
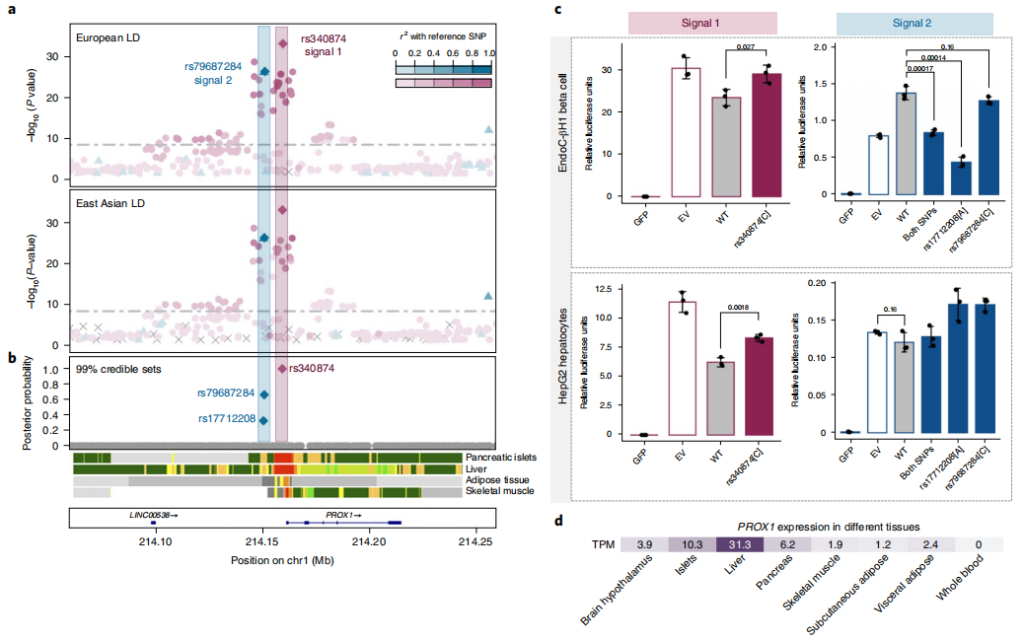
图6 PROX1位点分子机制
在DIAMANTE研究中,利用群体多样性来比较跨种族和祖先特异性T2D GRS的预测性能。每个祖先组中选择两个研究作为GWAS检验,使用可解释表型方差(pseudo R2)和每个风险评分单位的优势比(OR)评估GRS的预测性能。在排除检验GWAS后,研究人员重复了跨种族和祖先特异性荟萃分析,确定了lead SNVs位点达到全基因组显著性阈值(跨种族:P<5×10−9;祖先特异:P<5×10−8)。
在各自祖先群体GWAS检验中,祖先特异性GRS表现最好。然而,对于有效样本量最小的祖先群体(非洲人、西班牙人和南亚人),因为获得全基因组显著性的lead SNVs数量很少,祖先特异性GRS的预测能力很弱。对于这些代表人数不足的祖先群体,欧洲祖先特异性GRS表现更优,其原因:1、更多的lead SNVs;2、lead SNVs代表的T2D关联信号在祖先群体中大多是共享的。
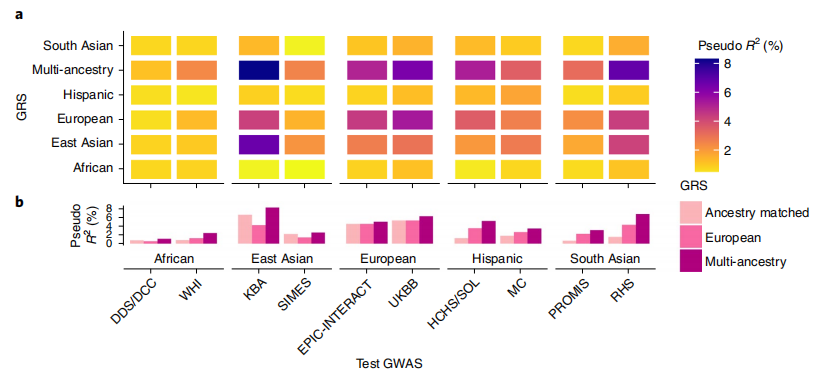
图7 跨种族和祖先特异性GRS在不同群体中的可转移性

图8 正向选择作用


