Nature子刊|多重碱基编辑技术实现人类基因组多个基因TAG转换为TAA
- boke
- 2022-11-10
- 7:33 上午
遗传密码具有简并性,除了3个终止密码子外,61个三联体密码子分配给20个不同的氨基酸,其中18个氨基酸由一个以上的同义密码子编码。研究人员发现,这20种天然氨基酸中的18种存在同义密码子(1种氨基酸有1个或多个密码子)编码。2013年,美国哈佛大学学者大胆地做了改造尝试,他们将大肠杆菌基因组上的终止密码子TAG替换成TAA,成功实现了同义密码子的替换。2019年,剑桥大学的科学家在《自然》发表了题为“Total synthesis of Escherichia coli with a recoded genome”的文章证明,通过在基因组范围内用定义的同义密码子替换目标密码子,用于编码标准氨基酸的密码子数目可以减少。
基于此,该研究团队通过高保真人工合成技术,将大肠杆菌4Mb的基因组替换为合成基因组,该大肠杆菌变体只有61个密码子,打破了传统生命体中64个密码子编码蛋白质的认知。2021年Science杂志上题为“Sense codon reassignment enables viral resistance and encoded polymer synthesis”的文章报道了Jason W. Chin 教授带领的团队,在全人工合成的大肠杆菌体内,通过敲除相关密码子TCG,TGA,TAG的tRNAs和释放因子,使该菌株实现了对噬菌体的完美抵抗力。同时将有意义密码子重新分配给非常规单体,实现了不同非常规单体的高效连续聚合以产生非常规杂聚物和大环化合物。
理论上,如果替换掉所有基因中的一个冗余密码子(即重新编码基因),并去除解码它的tRNA,人类细胞仍然可以制造它所有的蛋白质。但病毒的基因仍然包含这个冗余密码子,并且依赖宿主细胞机制进行复制,这样病毒将无法将它们的基因转化为蛋白质,使得试图复制的病毒被消灭,因此,基因组重编码的细胞将具有了免疫力。这种重新编码在人类基因组中的应用至今尚未有报道。
2022年8月2日,哈佛大学George Church教授团队、中国科学院深圳先进技术研究院合成生物学研究所刘陈立研究员团队合作在Nature Communications期刊发表了题为:Multiplex base editing to convert TAG into TAA codons in the human genome的研究论文。本研究初步证明了在人类基因组中将TAG转化为TAA的可行性,并为大型哺乳动物基因组工程提供了研究基础。GRIT软件可以开发成一种新的计算机辅助设计(CAD)平台,可以用于编辑更大的基因组。一旦完成,基因组重新编码的人类细胞将可广泛应用于生物医学,特别是开发对大多数自然病毒具有抵抗力的细胞疗法或治疗管线。
本文作者在本研究中作者提出了一个潜在方案来制备抗病毒人类细胞系:在全基因组范围内将终止密码子TAG转化为TAA,并将内源性真核释放因子1(eRF1)替换为具有选择性通读的工程化eRF1突变体,使得人类细胞系具有抗病毒的能力(图1a)。
作者选择终止密码子TAG的原因如下:(1)以前发表的论文报道,重新编码的大肠杆菌显示出非标准氨基酸的结合和多种病毒的抗性;(2)TAG是人类基因组中最不常用的密码子,允许较少的编辑;(3)理论上TAG可以用C-T碱基编辑(CBE)编辑成TAA,并增加gRNAs设计的灵活性,另外TAG是终止密码子,可以减少对CBEs引起的额外编辑对基因转录和翻译的影响。
结果
人类基因组编码软件GRIT的开发
考虑到人类细胞基因组水平重新编码的规模,需要能够自动执行设计的软件。为了满足这一需求,研究者开发了GRIT,它针对重新编码为基因组规模数据分析提供了一个定制化的Python分析流程(图1A)。GRIT的中心功能是解析基因组数据,寻找密码子TAG,并为具有PAMs序列特征的碱基编辑指南。
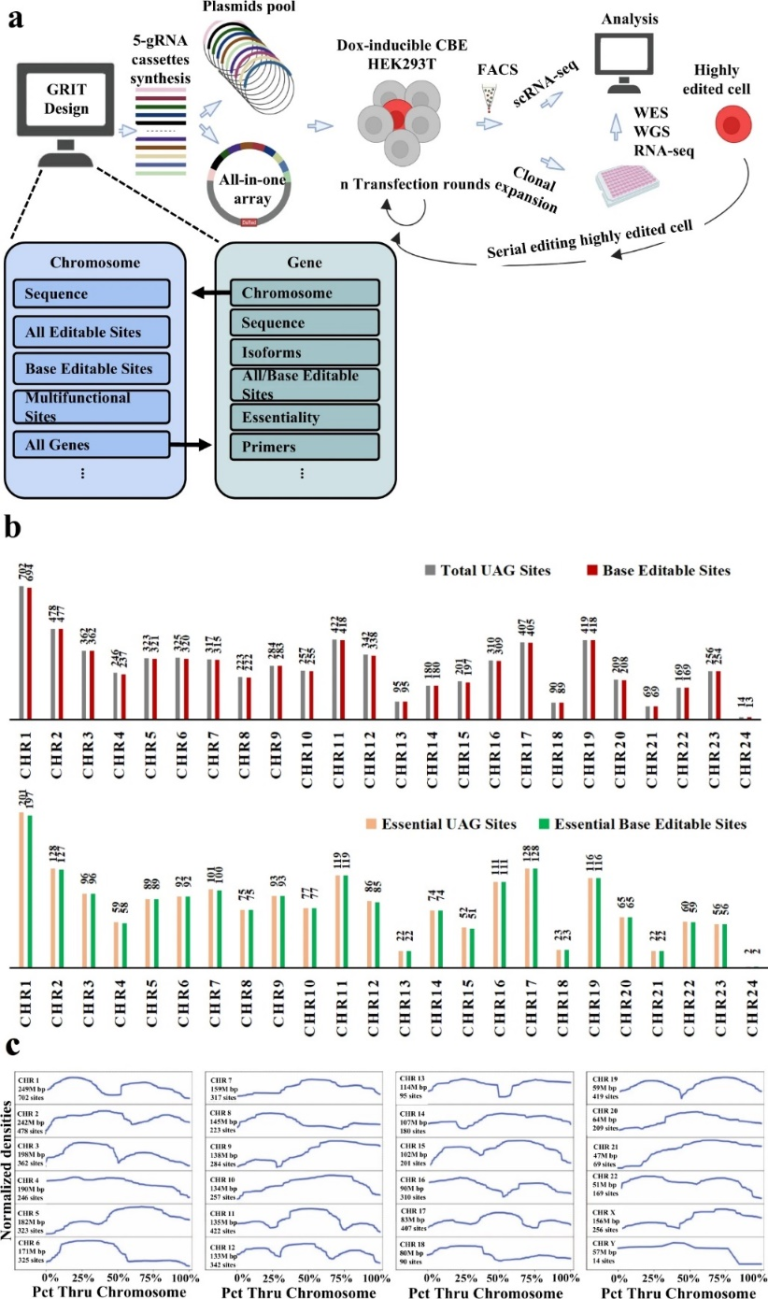
图1 用于将UAG转换为UAA以供人工重新编码的软件GRIT分析框架
GRIT为信息学分析提供了一个工具包,重点是重新编码。它的创建有三个关键的设计原则:(1)便携性:所有数据都可以下载,GRIT可以在台式计算机上安装并运行。(2)适应性:该项目的完整源代码在两个Python文件中,各种通用的和特定于重新编码的信息学数据随时可用,包括完整的基因和染色体序列。(3)易用性:GRIT带有预先编写的算法,用于复制结果和分析染色体数据、基因数据、标签位置数据以及编辑指南。特别是在重新编码时,GRIT可以用来标记基因组中的所有TAG位点,搜索可以用C碱基编辑程序直接编辑或用A和C编辑程序编辑的“菊花”序列标记位点,并设计相应的指南。
通过gRNA阵列在HEK293T细胞中进行多重碱基编辑
根据gRNA设计,CBE可以实现TAG到TAA的转换,但现有的多重基因编辑技术并不能满足需求,作者需要开发能同时将多个gRNA和碱基编辑蛋白送入单个哺乳动物细胞进行TAG到TAA重编码的技术。随着DNA合成能力的进步和成本的指数级下降,作者直接设计并合成了含有五个单独gRNA盒的gBlocks:五个先前发表的sgRNAs23(gBlock-PC)和五个设计的针对基因TAG区域的sgRNAs(gBlockYC1)(图2a)。将gBlock-PC和gBlock-YC1分别与evoAPOBEC1-BE4max-NG23共同瞬时转染到HEK293T细胞。Sanger测序和EditR31分析表明,来自gBlock-PC的sgRNAs的效率为~40-50%,略低于那些单独传递的相同sgRNAs23,而来自gBlock-YC1的sgRNAs的效率为~20-50%(图2b,c)。
然后,利用piggybac转座子系统产生了两个稳定的和多西环素诱导的HEK293T系分别与PiggybacFNLS-BE3-NG32和evoAPOBEC1-BE4max-NG(图2d)。作者分别瞬时将gBlock-PC和gBlock-YC1分别导入两个可诱导的CBE细胞系,gBlock-PC中sgRNAs的编辑效率在evoAPOBEC1-BE4max-NG细胞系中为~60-70%,略高于FNLS-Be3-NG细胞系中的~45-65%(图2e)。然而,来自gBlock-YC1的sgRNAs在evoAPOBEC1-BE4max-NG细胞系中的效率为~30-75%,显著高于FNLS-BE3-NG细胞系的~20-40%(图2f)。
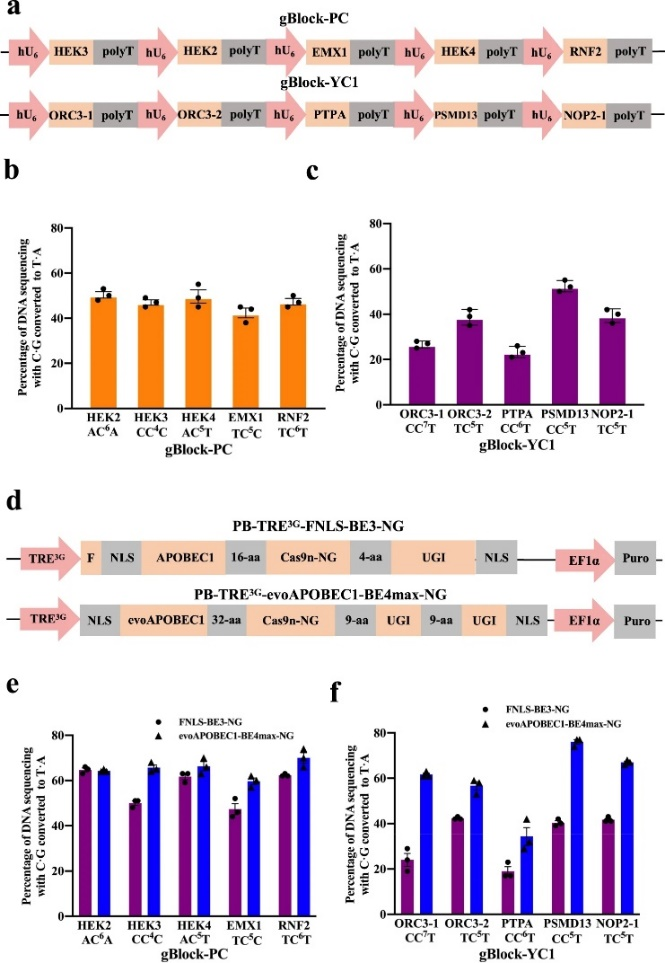
图2 利用gRNAs在HEK293T细胞中进行多重碱基编辑
用scRNAseq确定单细胞TAA标记重排
为了确定将TAG转换为TAA的有效策略,作者通过单细胞RNAseq进行突变检测。作者分离了约1000个单细胞,并进行了单细胞RNA-SEQ,以检查每个靶点在三个细胞群体中的分布(图1A)。作者总共绘制了38/52个基因位点,观察到在所有三种方法中,细胞数量都随着编辑位点数量的增加而减少,其中方法2(图3a)中编辑基因位点最多的细胞数量最多。作者绘制了细胞的种群密度图(图3b),并分析了每个目标和目标的编辑效率,编辑事件呈双峰分布(图3c)。还分析了每个单元格中每个映射站点的编辑效率(图3d)和每个样本中每个目标的总编辑效率(图3e)。总而言之,这些数据表明方法2是TAG到TAA替换的最有效的。
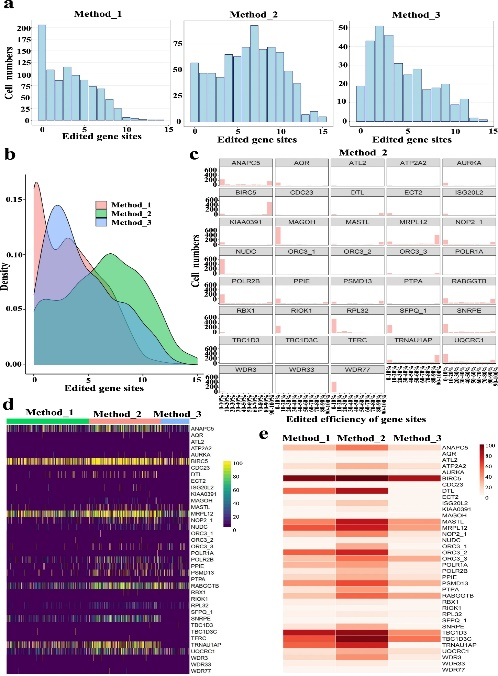
图3 将TAG转化为TAA的单细胞RNAseq实验策略评价
高度修饰的HEK293T克隆的培养和鉴定
为了进一步研究哪种方法产生高度修饰的可扩增克隆,作者从方法2和方法3的转染群中分选和培养单细胞,分别获得了28/96和24/96个单细胞克隆。对于方法2中的克隆,作者挑选了10个编辑良好的基因座(根据先前的WES测序分析,从每个gBlock中提取一个以验证其传递),进行PCR扩增,然后进行Sanger测序和Editr分析进行初步筛选。
结果表明,4个不含gBlock的克隆和24个具有1~10个不同gBlock数目的克隆,19个克隆包含全部10个gBlock。对于方法3中的克隆,作者使用10个编辑良好的基因座中的3个进行筛选,发现13个克隆没有编辑,11个克隆具有1到3个编辑位点,其中克隆#11、20、21和24具有全部3个编辑位点。然后,作者对2个高度修饰的克隆(克隆19和21)中的所有靶基因座进行了Sanger测序。在方法2的19个克隆中,作者观察到TAG到TAA的替换位于33/47个基因组位点,其中9个是纯合的,14/47个是未编辑的。来自方法3的克隆21显示了27/40个所需的编辑位点,其中10个是纯合子TAA,13/40个是未编辑的(图4a)。
这一结果与作者之前用scRNAseq检测到的结果一致。为了确定编辑效率是否可以随着随后的几轮转染而提高,作者还使用方法2将gBlock导入高度修饰的克隆19,并从22/96个克隆中选择了克隆19-1、19-16和1921,这是因为与原始克隆19相比,在选定的座位上进行了更高的编辑效率。
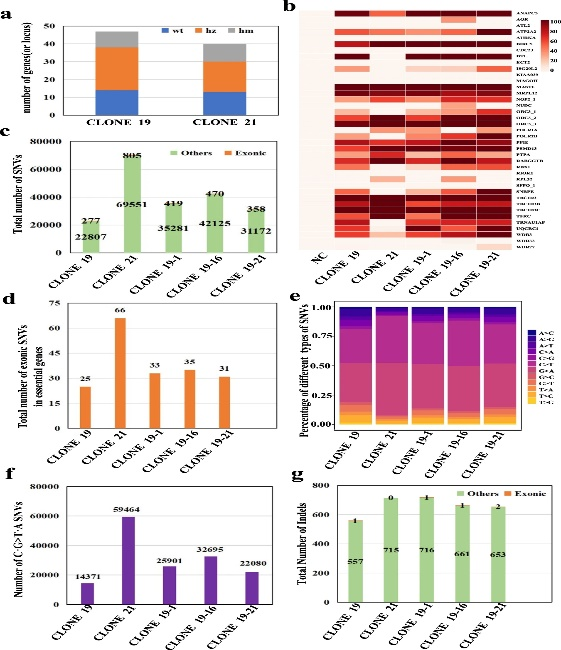
图4 WGS统计HEK293T高度修饰克隆的遗传变异
WGS对高度修饰的HEK293T克隆的中靶和脱靶效应分析
为了全面评估CBE全基因组TAG到TAA转化的中靶和脱靶率,作者对高度修饰的克隆(19,21,19-1,19-16,19-21)和阴性对照进行了30倍的全基因组测序(WGS)。对于靶点编辑,热图显示已经绘制了39/47个基因位点,其中25到28个在高度修饰的克隆中被编辑。这些可编辑网站的编辑效率范围从~33%到100%。与克隆19相比,克隆19-1、−16、−21在几个座位上的编辑效率提高了~10%至40%(图4b)。这一结果与作者之前用桑格测序检测到的结果一致。
为了发现脱靶事件,作者分析了高度修饰的克隆(19,21,19-1,19-16,19-21)中的单核苷酸变异(SNV)和插入/缺失(InDels),并与对照进行了比较。减去目标上的SNV后,SNV分别为23084、70356、35700、42595和31530(图4c)。对这些克隆的进一步分析发现,277、805、419、470、358个SNV分别位于外显子上(图4c),而仅有25、66、33、35、31个SNV分别位于必需基因的外显子(图4d)。作者将SNV分为单独的突变类型,发现C·G到T·A的突变是最频繁的编辑(图4e),克隆的C·G到T·A的SNV突变的数量分别为14371,59464,25901,32695,22080(图4f)。除SNV外,在这些克隆中分别检测到558、715、717、662、655个InDels,其中一小部分位于外显子(图4g),基本基因的外显子中没有。
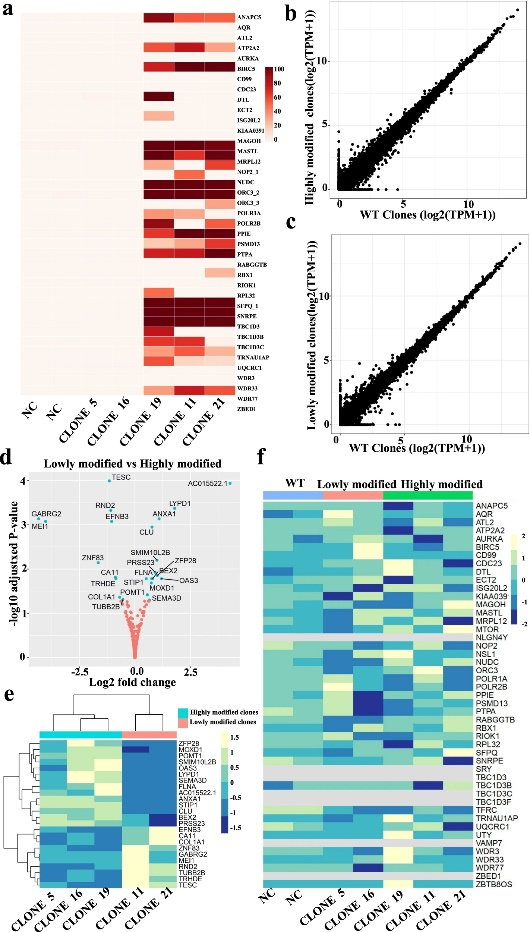
图5 通过混池RNAseq对高修饰HEK293T克隆和低修饰克隆进行靶向和基因表达分析
高度修饰的HEK293T克隆的基因表达和核型分析
为了检测高度修饰克隆编辑前后潜在的基因表达变化,作者对单细胞RNA-SEQ数据进行了UMAP分析,没有观察到大量编辑驱动的细胞聚集,表明编辑没有显著的基因表达变化。接下来,作者分析了高度修饰克隆(19、21和11)、低修饰克隆(5、16)和阴性对照的大量RNAseq数据。作者进行了OnTarget分析,结果与WGS的结果一致(图5a)。高修饰克隆和低修饰克隆的基因表达水平在所有基因(图5b-e)和43个靶基因座(图5f)中大多相似。一些基因在高、低和野生型阴性对照克隆之间差异表达(图5b),基因名称和基因表达倍数变化更详细地显示在图5e,f。作者还对高度修饰克隆和低修饰克隆之间的差异表达基因(DEG)进行了浓缩分析,没有发现任何基因集富集。
因此,混池rna-seq是一种有效的高通量多位点编辑筛选单克隆的方法,因为它的成本低于WES和WGS,而且还可以评估基因表达的基因编辑前后变化。作者还检查了多重基因组编辑是否发生了意外的基因组重排。对单个修饰克隆的核型分析表明,由于多重编辑,没有观察到基因组重排。
讨论
本文作者首次报道了HEK293T细胞系中多重碱基编辑将UAG转化为UAA密码子的GRIT分析软件。作者确定在6700个TAG中,6648个在人类单倍体基因组中是可编辑的。更重要的是,根据参考基因组版本GRCH38.p13显示,1947个持家基因其中1937个基因的TAG密码子是可编辑的。对于NG-CBE无法编辑的标签密码子,作者计划在未来使用Cas12a-CBE34和prime editor来编辑。虽然GRIT只在人类基因组数据上进行了测试,但它可以被用于其他已有高质量基因组数据的真核物种。
作者直接设计并合成了含有5个gRNA阵列的gBlock,其编辑效率与单个gRNA阵列相当,将其导入具有evoAPOBEC1BE4max-NG的稳定和多西环素诱导的HEK293T细胞系。作者还成功地将gBlock一次组装成一个含有43个gRNA的一体化质粒,这是一个含有比以前报道的37-39个sgRNA更多的阵列,并且大大减少了组装时间。然后,作者优化了一种单细胞RNAseq检测TAG到TAA重排的策略,这是第一次将其用于评估碱基编辑针对多个基因座的结果。作者一次批量编辑了47个目标位点中的33个。重复传递sgRNA可以提高编辑效率。这项工作证明了在人类基因组中将TAG密码子转换为TAA密码子的可行性,并建立了哺乳动物细胞中非重复基因座的多重基因编辑框架。
作者还观察到CBE介导的脱靶情况。当BE3和单个sgRNA分别在小鼠胚胎和水稻中过表达时,最初的CBE BE3会诱导全基因组随机突变,平均频率为5×10−8/BP和5.3×10−7/BP。对于数学假设期望的简单概算,在实验最好的克隆19中,为了在每个细胞的靶上获得9个纯合子重编码的基因终止密码子,在4764个必需基因中产生了额外24个杂合子脱靶突变。为了获得1937次精确的位点编辑,4764个脱靶基因(包括约1550个纯合基因)中发生5165次中靶(主要是从C到T)的泊松平均命中率。
如果能将脱靶率降低60倍,那么作者预计每个细胞<1个纯合子脱靶基因,因此,作者预测克隆没有纯合子脱靶基因。有些基因更难编辑,因此,基于CBE的编辑效率必须显著提高。腺嘌呤碱基编辑(ABE)具有较低的脱靶负担,但不适合重新编码标签。作者的团队之前记录了人类细胞中基于CBE的编辑的最高数量(~6300)在LINE-1重复元件中。然而,单一的引导RNA是为LINE-1元件中同源性最高的设计的,作者预测与多个gRNA相比,它产生的脱靶突变负担较低。在未来,脱靶效应可以通过较少脱靶的CBE、RNP和DddA-split碱基编辑而得到改善。
人类基因重新编码是一项系统和复杂的基因组工程。作者目前的研究是人类基因组重新编码的第一步,这为后续的努力打开了大门,比如大肠杆菌重组编码和Sc2.0合成基因组。虽然到目前为止,作者可以通过一次转基因在单个克隆中获得多达33个基因,但作者可以优化这个框架,以扩大到所有必需基因或所有以TAG终止的基因,这些基因可以转化为TAA。
在这里,作者提出了几种潜在的策略:(1)基于CBE突变体,Prime editor和DdCBE进一步开发新的具有低脱靶率、高编辑效率和PAM/Free的碱基编辑器;(2)通过在BAC或YAC载体和sgRNA库上使用更大的gRNA阵列来提高sgRNA的递送能力;(3)通过RNP和同步转染法等新的递送方法。人类重新编码路线图的下一个目标是以协调一致的方式利用所有策略和高度进化的编辑工具,进行低至一轮编辑,评估和优化重新编码效率,然后使用高度工程化的碱基编辑酶进行大规模的脱靶和旁观者突变清理。