


miRNA与罕见病
- boke
- 2024-11-28
- 6:03 下午
微小RNA (miRNA) 在罕见遗传疾病发病机制中的作用逐渐被了解。miRNA 是一类小的非编码RNA,通过沉默靶标信使RNA (mRNA) 来调控基因表达。其生物发生过程包括转录成pri-miRNA,经DROSHA-DGCR8复合体加工,转运到细胞质,并进一步经DICER加工,生成成熟miRNA。这些成熟miRNA整合到RISC中,在那里调控基因表达。
miRNA的失调与各种孟德尔遗传病和家族性疾病,包括DICER1综合征、神经发育障碍 (NDD) 以及与miRNA结合位点突变相关的疾病有关。例如,与MIR96突变相关的听力丧失、与MIR184突变相关的眼部疾病以及与MIR140突变相关的骨骼发育不良。
这些罕见疾病通常具有遗传基础,miRNA的失调可以通过影响关键基因的表达来促进其发病机制。miRNA生命周期涉及几个关键基因,包括DROSHA、DGCR8、DICER1和AGO1/2。DICER1基因的遗传变异被发现会导致肿瘤易感性疾病,通常称为DICER1综合征。过去两年,研究发现DGCR8中的胚系致病变异(GPV)与肿瘤易感性相关,而AGO1/2中的GPV与神经发育障碍(NDD)相关。
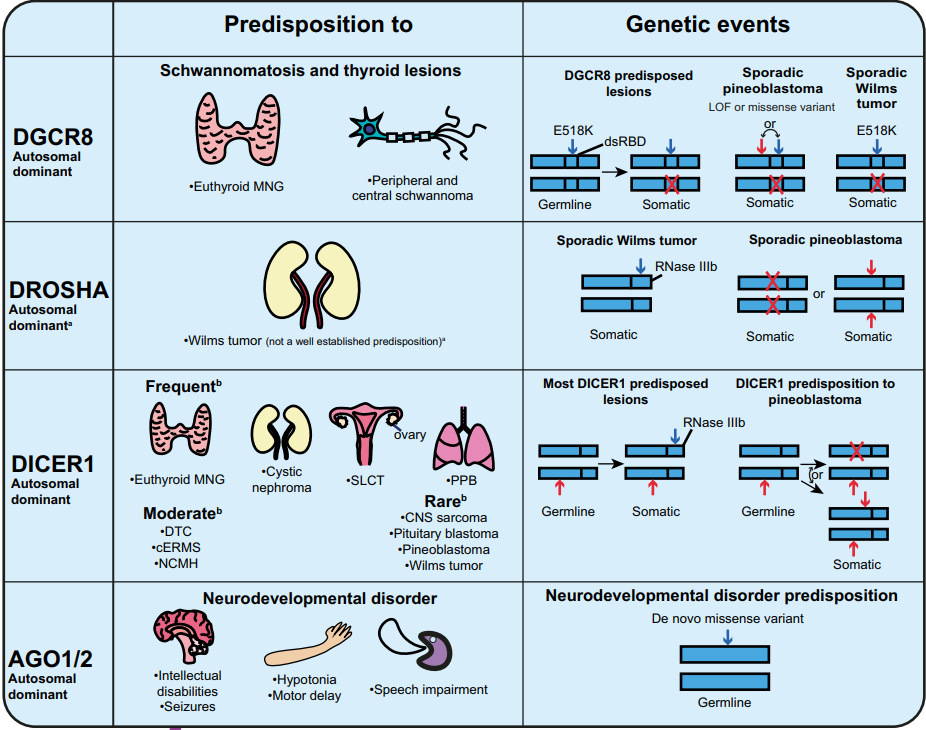
DROSHA和肿瘤易感性
DROSHA的体细胞突变常在肾母细胞瘤中发现。这些突变发生在DROSHA的RNase IIIb结构域中,会破坏金属离子结合和核酸内切酶活性,导致包括肿瘤抑制因子let-7家族在内的多种miRNA水平降低。肾母细胞瘤与胚系DROSHA变异R967W相关,表明此类突变可能使个体易患某些肿瘤。DROSHA的杂合缺失突变对miRNA产生影响很小。相反,DROSHA的RNase IIIb结构域的杂合突变表现出显性负效应,显着抑制let-7家族和其他miRNA的产生,同时仍允许足够的残余miRNA加工来支持肿瘤生长。DROSHA变异也已在两名患有严重智力障碍、癫痫、白质萎缩、小头畸形和畸形特征的个体中观察到。
DGCR8与甲状腺癌
DGCR8位于22q11.2染色体区域。在甲状腺癌中,DGCR8中反复出现的变异为c.1552G>A (p.E518K)。E518K变异导致成熟miRNA水平降低,与正常DGCR8相比,导致肿瘤中关键miRNA表达降低,这种降低与沉默DGCR8促进肿瘤生长的发现一致。此外,DGCR8 E518K突变已在散发性滤泡性甲状腺癌病例中观察到,其中额外的功能丧失(LOF)突变或杂合性丧失(LOH)似乎对于致癌作用是必要的。
DICER1综合征
DICER1综合征包括一系列罕见肿瘤,包括胸膜肺芽瘤(PPB)和囊性肾母细胞瘤,以及更常见的甲状腺功能正常的结节性甲状腺肿(MNG)。该综合征的GPV通常是功能丧失(LOF)变异,最常见的是无义突变或移码突变。为了使该综合征表现出来,DICER1基因中必须发生第二次遗传改变。第二次突变通常是错义变异,影响RNase IIIb结构域中关键残基(“热点”),这些残基对于金属离子结合和核酸内切酶活性至关重要。虽然不那么常见,但第二次打击也可能是剩余DICER1等位基因的杂合性丧失(LOH)。
Argonaute (AGO) 蛋白在神经发育障碍 (NDD) 中的作用
哺乳动物中,四种 Argonaute (AGO) 蛋白——AGO1、AGO2、AGO3 和 AGO4—是 miRNA 介导的基因调控的核心组成部分。这些蛋白是 RISC 的核心组成部分,它们结合单链 miRNA 并引导其到达靶标 mRNA。AGO1 和 AGO2 的基因变异与多种神经发育障碍 (NDD) 相关,包括智力障碍、发育迟缓和言语障碍,这些变异削弱了其通过miRNA调控基因表达的功能。
Lessel 等人发现,在患有 NDD 的患者中,AGO2中存在 13 种新的遗传变异,其中大部分是错义突变。功能实验表明,这些变异不同程度地削弱了AGO2的沉默功能,主要表现为功能缺失。值得注意的是,这些变异并未影响 AGO2 的剪切活性;相反,它们破坏了其与靶标 mRNA 分离的能力。这导致 mRNA 持续结合时间延长,并最终损害了基因调控。
类似地,Schalk 等人报告了 NDD 受试者中 AGO1 的各种基因变异,强调了它们对神经发育的影响。这些 AGO1 变异,包括错义突变和小缺失,与智力障碍和其他在 AGO 相关疾病中常见的临床特征相关。
最近的研究表明,AGO1 和 AGO2 可能参与 miRNA 靶向降解,该机制调节细胞内 miRNA 的周转和积累。AGO1 或 AGO2 变异导致 miRNA 水平失调,可能会导致基因表达模式改变,从而促成 NDD 的发病机制。
从临床角度来看,AGO1 和 AGO2 变异的鉴定扩展了我们对 NDD 基因基础的理解,突出了遗传检测在患有不明原因神经发育或心脏异常的患者中的重要性。这有助于医生根据受影响个体的特定基因谱提供更准确的诊断、个性化的治疗策略和家庭咨询。
MIR96 和耳聋
非综合征性耳聋 (NSHL) 是一种遗传性疾病,其特征是听力障碍,无其他症状,影响大约每 1000 名新生儿中 1 人,以及每 1000 名青少年中 3 人以上。
该疾病具有基因多样性,超过 120 种已识别的基因或基因座参与其中。与 NSHL 相关的特定基因座是 DFNA50,位于 7q32 染色体上。最初的测序并未在此基因座中发现编码基因中的致病突变。然而,对 miRNA 基因 MIR96 的全面遗传分析(该基因也位于该基因座内)发现了两个与西班牙两大家族中的 NSHL 相关的突变。这些突变表现出显性遗传。
MIR96 在耳蜗内毛细胞和外毛细胞中表达,对于它们的差异化至关重要,并在听觉后脑的发育中发挥作用。MIR96 中的突变会影响成熟 miRNA (miR-96-5p) 的种子序列,这对于识别和结合靶标 mRNA 至关重要。
西班牙家系:种子序列中的 n.13G>A 和 n.14C>A 突变会影响靶标识别。Luciferase reporter assay证实了这些突变 miRNA 对野生型 miRNA 靶标的沉默活性下降。这些突变还会影响 miRNA 前体结构的稳定性,导致 miR-96-5p 水平下降。

成熟的hsa-miR-96-5p和miR-96(+13G>A)和(+14C>A)突变已对齐。碱基“G”用黑色表示,突变碱基“A”用红色表示。
Solda 等人报道了一种新的 MIR96 基因变异,该变异在意大利家系中导致常染色体显性遗传的出生后进行性非综合征性耳聋。与之前的突变不同,n.57T>C 突变并不影响 miR-96 的种子区或成熟序列,但会影响 miR-96 前体的成熟,导致其成熟形式的水平降低。该突变预计会在 miR-96 发夹结构中产生一个凸起,可能干扰 DICER 加工。
MIR96 基因的突变是耳聋的罕见原因,但识别像 n.57T>C 这样的新变异可能有助于揭示 DFNA50 相关耳聋的潜在机制。n.57T>C 突变似乎导致 miR-96 水平降低,从而导致听力下降来得晚,而且症状没那么严重,与影响靶标识别的突变相比。受影响家系中听力丧失发作和进展的变异性,包括一些年轻个体的不完全外显率,表明不同的突变可能导致不同的致病机制。
在 diminuendo 小鼠中进行的研究,这些小鼠具有类似的 miR-96-5p 种子突变,显示出进行性听力丧失,伴随几种野生型靶标的上调,表明功能缺失效应。此外,这些小鼠与野生型相比显示出一些下调基因,暗示可能存在功能增强效应。MIR96 基因的突变会扰乱正常的 miRNA 加工和靶标调控,导致非综合征性耳聋。这些突变会引起 miRNA 前体结构的变化,导致成熟 miRNA 水平降低和基因调控改变。理解这些机制有助于深入了解耳聋的遗传基础和潜在的治疗靶点。
MIR184 在眼部疾病中的作用
在爱尔兰一个大家系里发现 miR-184 中的杂合 C-to-T 转换 (n.57C>T),该家族有常染色体显性遗传的早期发作的白内障和严重角膜圆锥症。同一突变还在一个被诊断为 EDICT 综合征(内皮营养不良、虹膜发育不良、先天白内障和基质变薄)的家系中发现。在这两种情况下,该突变都与疾病表型共分离,这突显了该突变在这类疾病中的致病作用。
角膜里含量最多的 miRNA 是 miR-184-3p,主要在角膜基底层和上皮层,以及晶状体中。它对角膜新生血管生成,以及表皮细胞的生长和分化,都非常重要。miR-184-3p 和 miR-205-5p 争夺结合位点,从而阻止 miR-205-5p 抑制其靶基因 INPPL1 和 ITGB4,两者对角膜基底上皮半桥粒正常工作和角质细胞凋亡的调控至关重要。miR-184 的杂合 C-to-T 突变 (n.57C>T) 发生在成熟 miR-184-3p 的功能性七个碱基 miRNA 种子区 (GGACGG) 的中心位置。这会破坏与 miR-205-5p 的竞争,从而允许 miR-205-5p 下调 INPPL1 和 ITGB4,这有助于眼部疾病的病理发生。
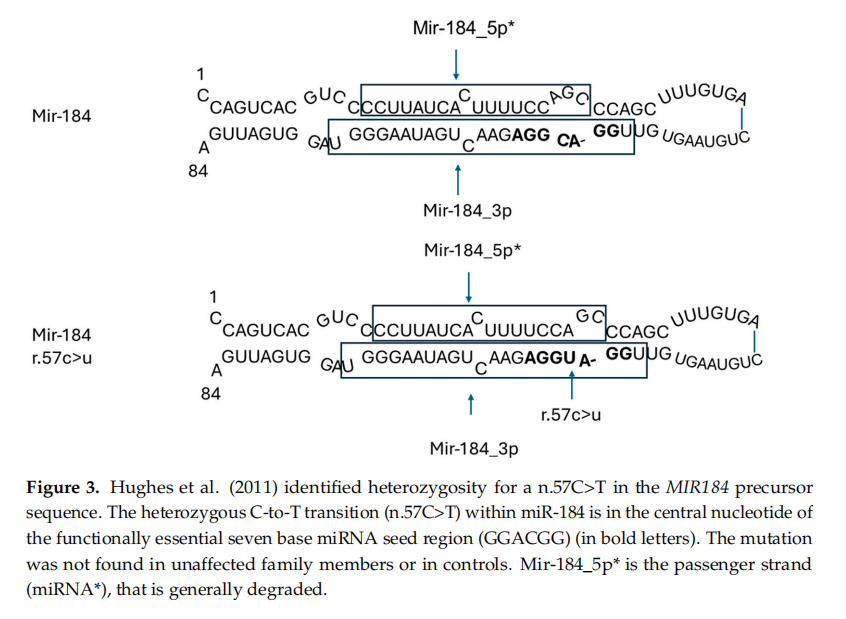
Hughes 等 (2011) 发现 MIR184 前体序列中存在 n.57C>T 杂合突变。miR-184 中的 n.57C>T 杂合突变位于功能关键的七个碱基 miRNA 种子区 (GGACGG),(粗体)位于中心核苷酸位置。该突变未在无症状家族成员或对照组中检测到。miR-184 5p* 是乘客链 (miRNA*),通常被降解。
MIR184基因(n.3A>G和n.8C>A)的两个突变体在受孤立性角膜圆锥症影响的家系中被发现。这两个突变体,miR-184 (n.3A>G)和miR-184 (n.8C>A),破坏了miR-184的茎环二级结构。miRNA前体结构的完整性对于DROSHA和DICER酶(它们对miRNA加工至关重要)的识别和切割至关重要。这些结构变化可能会干扰miRNA的成熟过程,进而影响miRNA的表达,并改变下游的生物学途径和通路。
MIR204与遗传性视网膜变性
遗传性视网膜变性 (IRD) 是一类由光感受器功能障碍或退化引起的疾病,通常会导致视力障碍或失明。在英国一个五代家系中,miRNA 基因突变被发现与 IRD 相关,其特征包括遗传性视网膜变性、视神经裂孔闭合缺陷和虹膜缺损等重叠表型。大量研究表明,miR-204 在其表达的眼部组织的发育和功能中发挥着关键作用。体外研究表明,miR-204 可能参与视网膜色素上皮细胞的正常分化和功能。
在斑马鱼实验中,敲低 miR-204 的表达会导致光感受器逐渐受损并死亡。miR-204-5p 在视网膜色素上皮 (RPE) 和睫状体中表达较高,而在视网膜内核层和脉络膜中表达较低。它在视网膜发育和功能中起着关键作用,可能为光感受器提供神经保护。n.37C>T 突变并未改变 miR-204-5p 前体或成熟形式的表达水平,这意味着它不影响 miRNA 的正常产生。然而,该突变发生在 5p 臂的种子区,这会影响靶基因的识别。这可能通过两种机制发生:一是bona fide miR-204 靶标识别受损(功能缺失);二是产生新的异常靶位点(功能获得)。
miR-204 位于TRP通道基因TRPM3的第 8 内含子,位于 9q21.12 染色体区域。9q21 内以及特异性包含 TRPM3 基因的缺失并未被报道会引起眼部表型,而是会导致智力迟缓、癫痫、言语发育迟缓、自闭症行为和中度面部畸形。该突变 (n.37C>T) 位于 miR-204-5p 种子序列的第四位。该突变在受影响的家族成员中呈常染色体显性遗传,并与疾病表型完全符合,这表明 n.37C>T 突变通过功能获得机制起作用。后续研究需要识别突变 miRNA 的具体靶点,并探索针对 IRD 中 miR-204-5p 调节的潜在治疗方法。
MIR140与骨骼发育不良
骨骼发育过程中基因表达异常可能导致骨骼发育异常、软骨异常、骨骼结构异常以及关节异常。骨骼发育不良是一组高度异质的遗传疾病,影响软骨和骨骼的生长发育。这些疾病是由各种基因突变引起的,包括miRNA基因MIR140。miR-140是研究最为深入的与软骨发育相关的miRNA之一。根据斑马鱼和老鼠胚胎的原位杂交研究,miR-140主要在软骨中表达,而在其他组织中表达较少。miR-140-3p在软骨中含量显著高于miR-140-5p,约高出10倍。miR-140-3p和miR-140-5p都在软骨发育和稳态中发挥作用。敲除小鼠实验表明,miR-140-3p对维持软骨功能至关重要,其缺失会导致轻微的骨骼发育异常。
在两大家系中,通过一项专注于极罕见先天性骨骼疾病的分子诊断项目,发现了一种新的骨骼发育不良。临床表型包括比例失调的矮小身材、四肢变短、手足变小以及中面部发育不全,鼻部异常小。影像学检查显示轻微的脊柱发育异常、髋关节和膝关节的骺板发育延迟以及严重的指骨短缩,指骨骺锥形。这些骨骼异常在成年后可能发展为早发性脊柱炎和退行性关节病。所有受影响个体均具有正常的认知功能、牙齿、听力和视力。全基因组测序在两大家系中均检测到相同的杂合核苷酸替换(chr16:69967007A>G,MIR140: n.24A>G)。该变异影响了高度保守的miRNA miR-140-5p(由MIR140基因编码)的种子序列第一个核苷酸。
该突变突出了miRNA在骨骼发育和稳态中的重要作用。为了开发针对骨骼发育不良疾病的靶向疗法,深入了解这种突变所破坏的特定靶标和分子通路至关重要。
MIR17HG:Feingold Syndrome Type 2
Feingold Syndrome是一种常染色体显性遗传疾病,特征是学习困难、身材矮小、小头症和指骨发育不良。MYCN基因的致病基因变异是费因戈德综合征的主要原因;一些没有MYCN突变的病例与13q31-q32区微缺失有关,特别是影响MIR17HG,它编码miR-17-92簇。
全基因组比较基因组杂交分析显示,在患有类似Feingold Syndrom骨骼异常且无MYCN突变的患者中,13q31-q32区微缺失范围为165 kb至17 Mb。这些微缺失在受影响的家族中与疾病表型相关,表明MIR17HG的单倍体不足是根本原因。这些缺失的最小重叠区域包含MIR17HG,从而将这些病例归类为Feingold Syndrome Type 2 (FGLDS2)。
MIR17HG编码miR-17-92簇,也称为癌基因miRNA-1,因为它在多种癌症中具有致癌作用。在正常细胞中,该miRNA簇对肺、心脏、骨骼和免疫系统发育至关重要。由于这些微缺失,MIR17HG丢失一个拷贝,导致单倍体不足,从而扰乱miR-17-92簇调节的正常发育过程。相反,在与MIR17HG重叠的13q31区观察到微重复,这些患者表现为身材高大、头大、发育迟缓和骨骼异常,这表明由于微重复导致的miR-17-92簇过表达会导致不同的临床表现。
MIR9-3和MIR1299的拷贝数变异与先天性肾脏和泌尿道异常(CAKUTs)
CAKUTs是影响约每500名新生儿中1人的结构和功能异常,是儿童肾衰竭的主要原因。包括点突变和罕见拷贝数变异(rCNVs)的遗传因素占CAKUT病例的20-25%。值得注意的是,miRNA基因在与CAKUTs相关的rCNVs和常见CNVs中较为丰富。这些区域中的miRNA调节关键的细胞过程,例如生长、分化、炎症和凋亡,这些对于CAKUT发育至关重要。涉及MIR9-3和MIR1299的拷贝数变异与CAKUTs显示出显着关联。尽管miRNA已被提议作为基因型-表型研究中的潜在遗传因素,但它们在CAKUTs中的具体作用仍不清楚。
通过削弱免疫系统平衡,从而导致疾病的发生和发展。miR-29 因为能抑制胶原蛋白和转化生长因子-β (TGF-β) 等促纤维化基因的表达而知名。
HDAC6 基因的 miR-433 结合位点突变与 X 连锁显性软骨发育不良症有关
与软骨发育不良症相关的基因编码细胞外基质蛋白、信号分子、转录因子或代谢途径的成分。一个 X 连锁显性软骨发育不良症家系被发现携带一个新的变异,c.*281A>T,位于 HDAC6 基因的 3′ 非翻译区 (UTR),该变异与该家族的疾病密切相关。该突变破坏了 miR-433 对 HDAC6 的转录后调控,导致受影响个体 HDAC6 表达量增加。这种 HDAC6 失调对软骨和骨骼发育有很大影响,因为 HDAC6 影响细胞分化、细胞骨架组织和蛋白质代谢等过程。
c.*281A>T 变异位于与 miR-433 种子序列相对应的序列中,预测的 HDAC6 3’UTR 中的 miR-433 位点包含 miRNA 种子序列 (miRNA 核苷酸 2-7;5’UCAUGAUG3′)。miRNA 核苷酸2-7的完美连续匹配对 miRNA 的生物学和功能至关重要,任何错配都会显著影响基因表达调控。c.*281A>T 变异将预测的 miRNA 位点从 5’AUCAUGA3′ 改变为 5’AUCUUGA3’,从而破坏miR-433的结合,导致 HDAC6 表达增加。

X连锁显性软骨发育不良患者中HDAC6基因3’非翻译区(3′-UTR)的变异体
下表罗列了与miRNA相关的基因及其相关的罕见病。

随着基因测序技术的进步,很多研究者开发了数据库来存储关于miRNA与疾病的数据。这些数据库大大受益于计算生物学,提供了宝贵的数据。关键数据库包括:HMDD:miRNA与疾病数据库 (http://www.cuilab.cn/hmdd);dbDEMC:癌症miRNA表达差异数据库 (https://www.biosino.org/dbDEMC);miR2Disease:miRNA和疾病数据库 (http://www.mir2disease.org/);miRCancer:miRNA和癌症数据库 (http://mircancer.ecu.edu/) 。这些资源通常用来预测miRNA与疾病的关联。
结论
关于参与miRNA生物发生、加工和基因表达过程的基因中的致病变异,相关报道不多。miRNA在基因表达调控方面至关重要,并日益被认为是各种遗传疾病的重要因素。它们能够靶向多个mRNA并影响广泛的细胞过程,突显了它们在正常生理和疾病病理学中的复杂性和重要性。miRNA基因及其调控通路突变可能导致关键基因失调,从而导致神经发育障碍等遗传疾病。
越来越多的证据表明miRNA与遗传疾病有关,强调了理解这些疾病的分子机制,识别miRNA-mRNA相互作用至关重要。测序技术和生物信息学工具的进步,使得研究miRNA靶标的全貌成为可能,从而为所涉及的病理机制提供了宝贵的见解。但要完全理解miRNA在疾病发病机制中的作用,仍有许多挑战。
总而言之,miRNA研究在加深我们对遗传疾病的理解,通过创新诊断和治疗方法,在改善患者预后方面具有巨大的潜力
参考资料
1.MicroRNA and Rare Human Diseases. Genes (Basel). 2024 Sep 25;15(10):1243.
2.miRNA biogenesis and inherited disorders: clinico-molecular insights. Trends Genet. 2023 May;39(5):401-414.
推荐阅读
这是示例文本,单击 “编辑” 按钮更改此文本。