结构变异(SV)是人类基因组中常见的遗传变异,可能导致不同的表型和疾病,包括癌症。然而,常用的二代测序读长较短,对结构变异的检测有限,不利于我们了解结构变异。在这项研究中,北京大学人民医院和北京希望组等机构的研究者们开发了28-Gene全长Panel(伯科生物合成),并利用Oxford Nanopore Technologies和 Pacific Biosciences长读长测序平台(TGS),对19名乳腺癌患者的肿瘤、癌旁组织和血液样本进行长读长基因组和转录组测序,分析了乳腺癌相关28个基因的结构变异。本研究结果表明,一些体细胞的结构变异在选定的基因中反复出现,并且大多数发生在非外显子区域。研究发现不仅有单核苷酸相关的热点区域,也有结构变异反复出现的热点区域。本研究通过采用靶向长读长测序来识别乳腺癌患者的SV,并证明了该方法在临床应用中有巨大潜力。
研究背景
乳腺癌是女性中最常见的恶性肿瘤。基因组的不稳定性是乳腺癌的一个关键分子特征,而结构变异直接表现出基因组的不稳定性。结构变异(SVs),包括插入、删除、复制、倒位和易位,相比较单核苷酸变异(SNVs)对基因组的影响程度,结构变异产生更大改变。在千人基因组项目研究中发现,结构变异是一种常见变异,已知有些特定的变异会导致遗传疾病和癌症。以往关于结构变异对基因结构和表达的影响的研究,大大加深了我们对肿瘤发生的认识。许多致癌基因已被证明是染色体易位的产物,并可作为治疗的靶点。然而,由于NGS测序读长短、PCR扩增偏好性的限制,在肿瘤基因组中识别复杂的SVs仍然具有挑战性。即使有研究开发相应的算法,仍然无法避免较高的假阴性率。第三代测序技术包含PacBio的单分子实时测序和ONT的牛津纳米孔长读长单分子测序技术。两种技术在检测结构变异方面都表现出较高的敏感性和特异性,已被应用在乳腺癌研究。
尽管乳腺癌在SKBR-3细胞系和患者来源的类器官中的SVs已被广泛研究,但仍然需要更多的证据来说明SVs和癌症之间的关系。长读长测序技术已经在癌症研究中显示出了它们的优势,这使得我们能够以前所未有的精度分析单倍体基因组。它们可以为精准医学提供有价值的见解,例如与单一热点突变相比,PIK3CA顺式双突变的患者对PI3K抑制剂阿培利司(alpesilib)表现出更高的敏感性。
本研究通过第三代测序平台(ONT和PacBio)以及靶向测序技术(28-Gene三代测序Panel),对肿瘤组织、癌旁组织和相应的血液样本进行长读长测序,准确检测乳腺癌基因组的结构变异。
研究路线
病例样本的选择
本研究选择多种乳腺癌亚型作为研究对象,包括4种侵袭性亚型(LuminalA、LuminalB、HER-2富集和三阴性乳腺癌(TNBC))和导管原位癌(DCIS)病例。每个病例的样本包含血液、癌组织和癌旁组织,以及7例健康捐献者的血液样本。
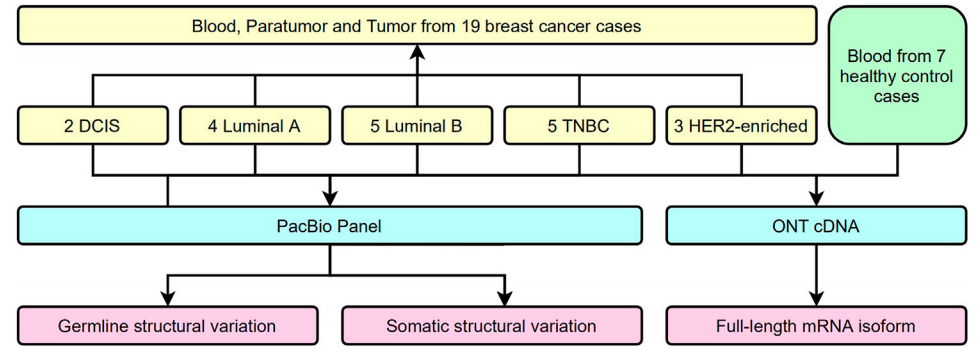
图1 研究设计流程图
Panel设计
本研究中Panel基因的选择有两种类型,包含参与了同源重组修复(HRR)的20个与乳腺癌高危人群相关的基因,以及8个与乳腺癌精准治疗相关基因。探针设计覆盖这些基因的全基因组区域,排除了重复序列部分,Panel约5Mb。
Panel性能
靶向捕获Panel在3种类型样本的捕获效率、目标覆盖率等基本指标中没有显著差异,患者与健康样本间也无明显差异。均能达到满足分析需求的测序深度,序列读长N50 约为3500个碱基,目标基因覆盖率>99.5%。
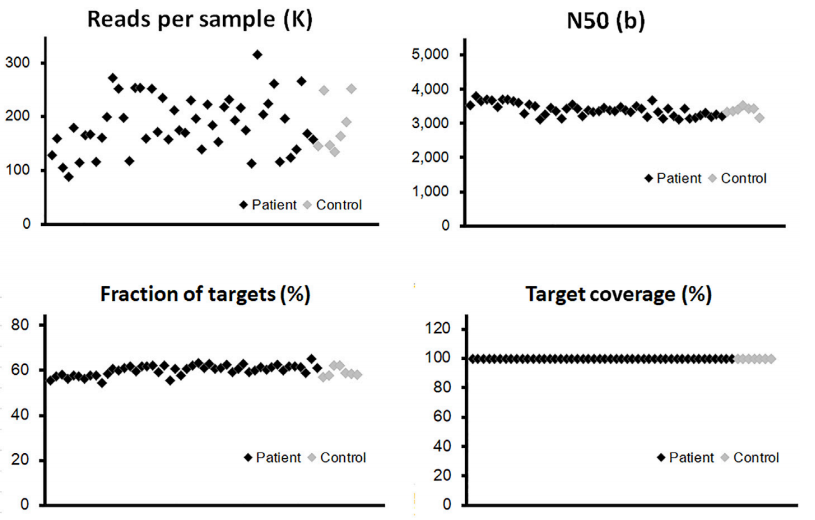
图2 28-Gene三代测序Panel数据表现
实验结果
乳腺癌患者胚系SVs的分析

图3A 特定基因和患者的胚系SVs特征
在本研究中,在12例患者(12/19,63%)的血液样本中检测到胚系SVs,每个患者大约携带1~6个变异。根据变异发生的基因组位置,这些变异分布在外显子、内含子、基因的上下游区域5’或3’非翻译区,以及基因2kb的侧翼区域,有些变异涉及不止一个区域。只有很少的变异发生在外显子区域,约占6/33,图中用亮蓝色表示。从变异所在基因类型看,既出现在HRD基因上,也会发生在与治疗相关的基因。这些变异呈散发状态,没有明显的聚集,推测与样本数量较少有关。

图3B EGFR基因的结构变异
值得注意的是,长读长测序方法允许我们在传统短读长测序技术难以分析的位置检测到SVs。例如,在患者RM65B中发现了在EGFR的3’UTR处约250个碱基的插入,但在健康对照组RMH3中没有。这个UTR区域靠近7号染色体的着丝粒,包含许多TA重复序列。同时,在该个体reads分布中可观测到该插入的位置和大小略有不同,如图3B所示,这进一步说明了该突变位点的复杂性。
肿瘤组织中体细胞SVs的潜在热点
体细胞SVs可以通过对比肿瘤组织中独特的SVs或血液样本或非肿瘤性组织中的SVs来识别。通过对比癌旁组织和匹配的血液样本的SVs时,发现很多共同变异,意味着这些常见的SVs可能是背景胚系变异。同时,在癌旁组织和血液样本中发现的独特的SVs,有可能来自杂合性丢失(LOH)。这也表明癌旁组织虽然在组织形态上是正常的,但是由于发生了SVs,说明在遗传学上已经有了改变。这一发现与之前SNV研究一致。
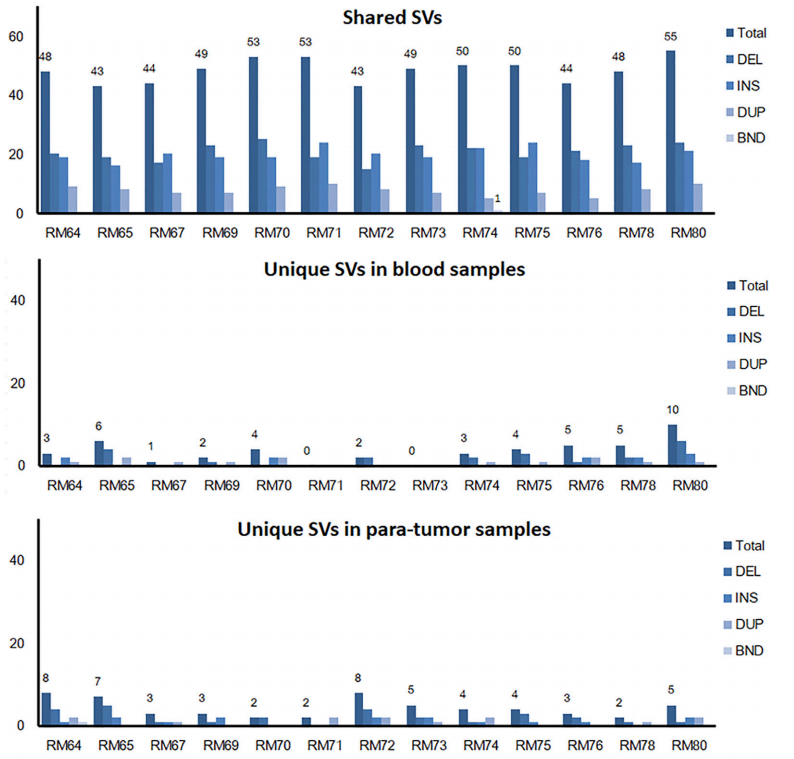
图4 癌旁组织和血液样本中共同和特有的SVs
本研究通过配对的血液样本为参照,挖掘肿瘤组织的肿瘤驱动SVs。28个乳腺癌相关基因的体细胞SVs分布情况如图5。每个患者只携带很少的SVs,或没有SVs。28个基因中,有12个检测到SVs,占比43%,SVs倾向分布在内含子。在这12个基因中,ERBB2在4个患者中出现,且都位于内含子区域,具有最高的SVs发生频率,另外两个频率较高的基因是NF1和RAD51B。
图5B总结了ERBB2在4个患者中SVs的情况:两个插入和两个重复。其中三个发生的位置邻近,但是这些患者在临床被分在三个不同的临床分型。这一区域AT富集,尚未有文献报道与疾病相关。然而,在ERBB2基因中体细胞SVs的相对富集(19例独立患者中的3例)表明,此区域可能与乳腺癌相关,但是需要更多的样本做进一步验证。
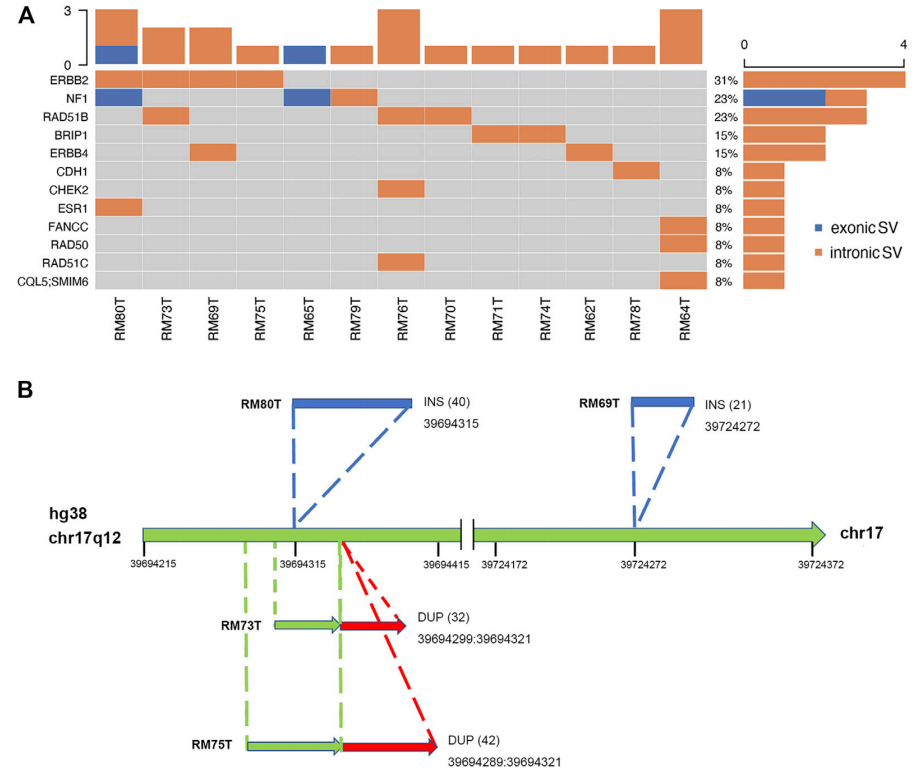
图5 非外显子区的体细胞SVs分布
肿瘤和癌旁组织的全长转录组分析
转录水平上的变化可以为基因组突变提供支持和直接证据。通过Nanopore的PromethION平台对肿瘤组织和癌旁组织的全长转录组测序。测序得到的序列长度均值为1.3kb,长度中位数为1.9kb。主成分分析可以清楚地区分两种组织来源的转录组数据。reads密度图(RPG10K)显示,肿瘤组织中基因的reads明显短于癌旁组织,但是由于样本量小,肿瘤组织的reads比癌旁组织短的结论尚不明确。综合上述特征,转录组长读长测序可能是未来诊断应用的候选技术。
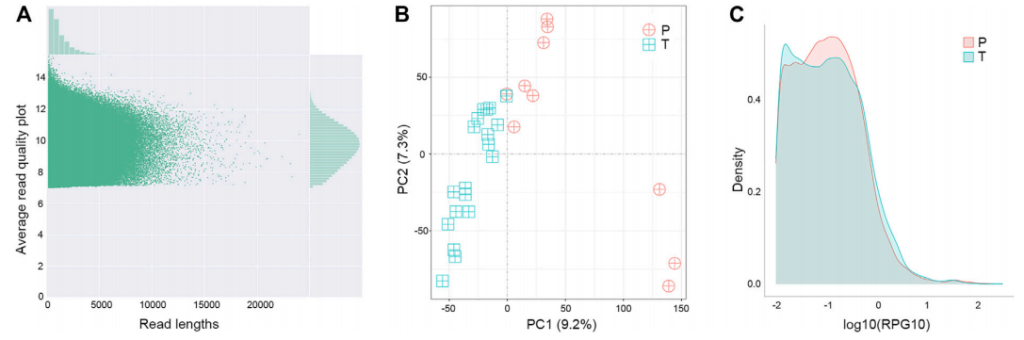
图6 肿瘤样本与非肿瘤样本的转录组序列长度差异
具有基因组学和转录组学证据的基因融合
融合基因的累积是肿瘤组织的常见模式之一。然而,关于融合基因是如何在癌症进展过程中促进或形成的研究比较少。PacBio和ONT平台长读长测序的优势有利于融合基因的研究。在19个病例中,研究者在6个患者中发现7个融合基因现象。在RM64病例中,位于chr17染色体的RECOL5基因,与chr8和chr7染色体的两个片段发生基因融合。高保真PacBio HiFi平台的高覆盖率reads(深度>30X,图7B)和ONT平台的转录本证据(图7C)均支持了该融合基因的可信度。
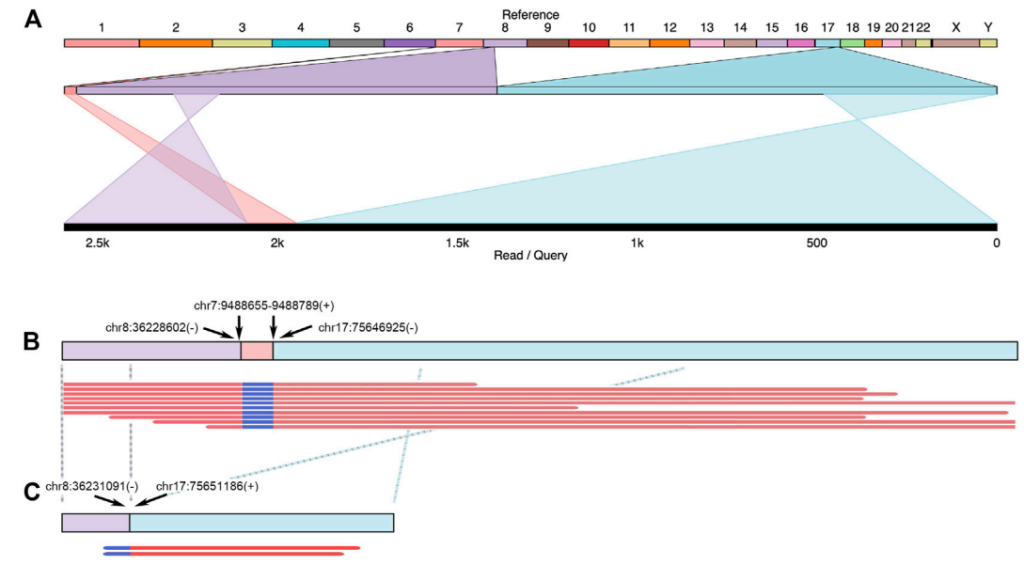
图7 在患者RM64的肿瘤组织中的融合基因RECOL5
讨论
已有很多研究证实PacBio长读长测序可以检测人基因组中超过20Kb的SVs,但是由于对全基因组进行三代测序成本太高,临床应用比较困难。为了解决这个问题,本文研究人员开创性地设计了适用于长读长测序平台的靶向捕获Panel,并成功应用于临床乳腺癌样本研究。通过基因组和转录组长读长测序对融合基因的研究表明,28-Gene三代测序Panel能够准确、稳健地鉴定SVs变异,表明基于三代测序Panel的长读长靶向捕获测序方法是一种有效检测癌症相关SVs的技术路线,在未来的临床研究与应用转化中具有巨大潜力。
值得一提的是,28-Gene三代测序Panel由伯科生物高品质核酸合成平台提供。不论是组织还是血液样本,该Panel的靶向测序表现优异,呈现出中靶率高,高覆盖率(图8),同时,在55例样本中表现出高度的性能稳定性。
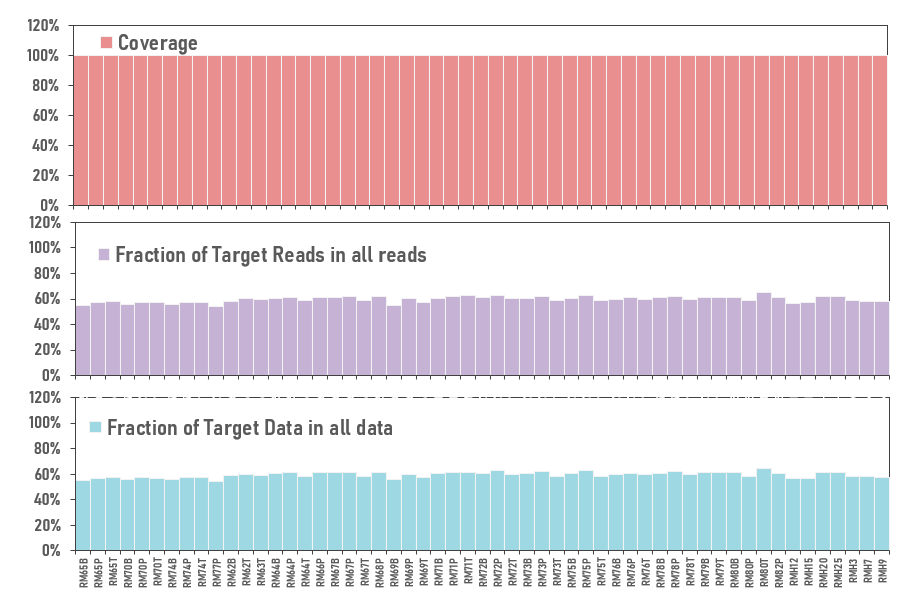
图8 28-Gene三代测序Panel主要性能表现(Table S2)