


Genome Research | 长读长适应性采样靶向测序与短读长全基因组测序用于小脑共济失调遗传诊断的对比研究
- boke
- 2025-03-19
- 3:50 下午

小脑性共济失调症 (CAs) 是一组由进行性共济失调为特征的异质性疾病。
已鉴定出17个重复扩展 (Repeat Expansion,RE) 位点作为主要遗传原因,占遗传诊断比例超过80%。尽管如此,诊断检测仍然有限且效率低下,通常采用单基因检测法。
本研究评估了长读长适应性采样靶向测序(LR-AS)和短读长全基因组测序(SR-GS)在CA诊断中的有效性。
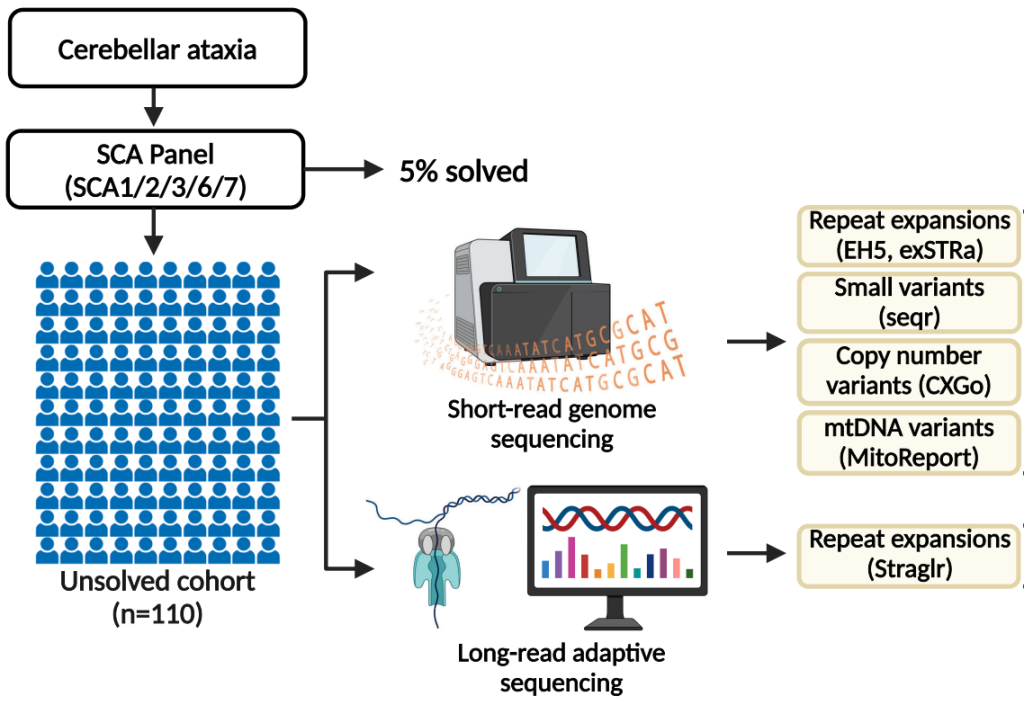
我们招募了110名 (女性 48 人,男性 62 人) 临床诊断为小脑性共济失调的患者。短读长测序 (SR-GS)用以鉴定致病性重复扩展(RE)以及CA相关基因的非RE变异。长读长Nanopore适应性采样靶向测序(LR-AS)用于识别重复扩展(RE)。
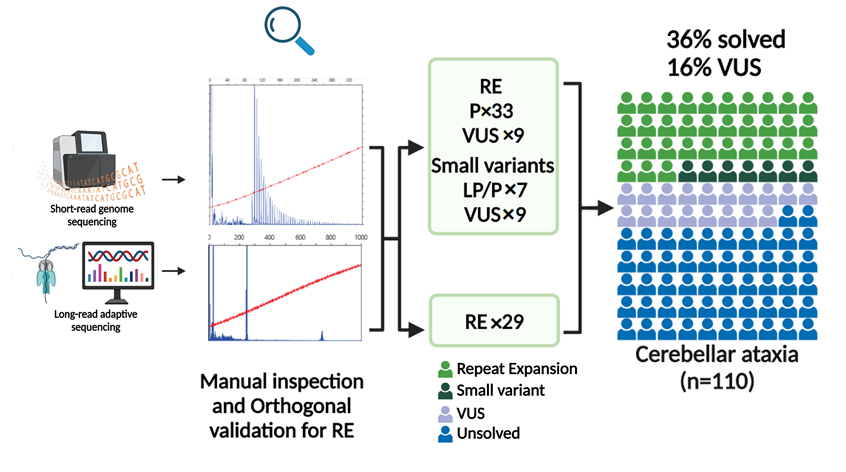
短读长测序(SR-GS)为队列中38% (40/110) 的患者提供了遗传诊断,包括7个非重复扩展(RE)致病变异。
重复扩展(RE)共导致33人患病,最常见的情况是SCA27B (n = 24)。相比之下,长读长测序(LR-AS)识别出29名患者患有致病性重复扩展(RE)。
两种方法对重复扩展(RE)的识别结果基本一致,但LR-AS未检测到4例SCA27B,原因是读数深度不足。
两种技术都受益于对重复扩展(RE)比对的手动复核,从而提高诊断准确性。
针对SCA27B的独立测试显示,短读长测序(SR-GS)的假阳性率为15%,长读长测序(LR-AS)为0%。
总而言之,这两种技术都是小脑性共济失调症(CAs)的强大筛选工具。短读长测序(SR-GS)技术已较为成熟,诊断机构已广泛使用,只需略微调整生物信息学流程即可用于CA诊断。LR-AS 在RE检测和表征方面优势明显,但需进一步优化才能在临床中更好的使用。
短读长测序(SR-GS)鉴定重复扩展(RE)
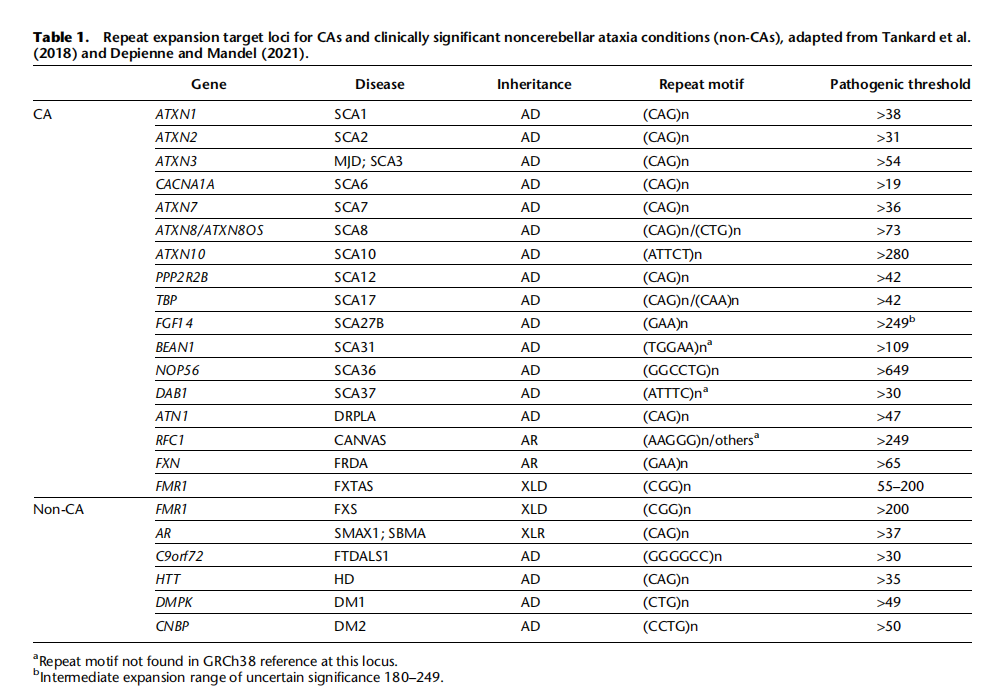
共有 67种已知的致病性RE,其中17种与CA直接相关,其余则与更广泛的神经遗传、神经肌肉及其他健康问题相关。鉴于研究队列的构成和纳入标准是CA,分析重点是表1中列出的22种 RE。
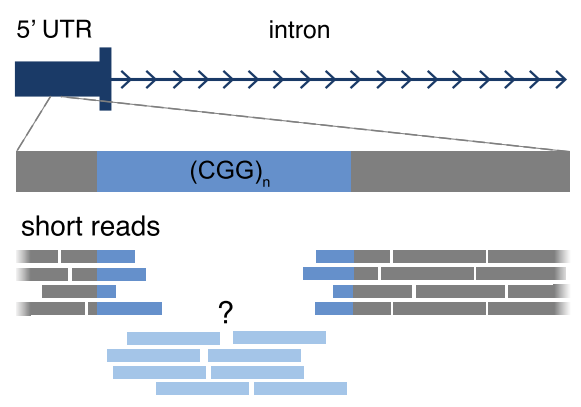
当DNA重复长度与标准SR-GS读长(150 bp)相近或更短时,短读长测序(SR-GS)在鉴定和测定致病性RE方面具有高灵敏度和高特异性。但对于较大的致病性RE,短读长测序(SR-GS) 会明显低估致病等位基因的大小。
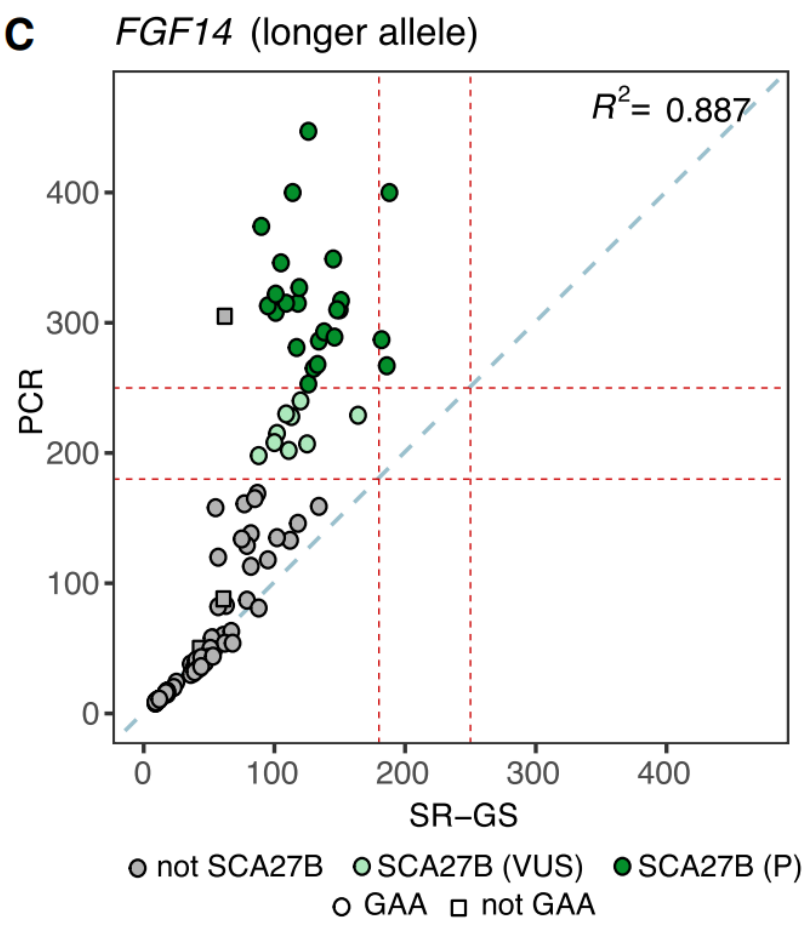
先前的研究还证明,当FGF14中导致SCA27B的RE扩张超过约 (GAA)100时,RE的大小估计不可靠,这表明SR-GS可能对该RE的特异性和/或灵敏性较差。
在该研究中,采用短读长测序(SR-GS) 检测的FGF14 GAA ≥90重复作为SCA27B疑似患者的判定阈值。与FGF14基因座的长程PCR测定的结果相比,SR-GS在鉴定FGF14中的致病性RE时具有100%的灵敏度和85%的特异性。
短读长测序(SR-GS) 鉴定非RE致病变异
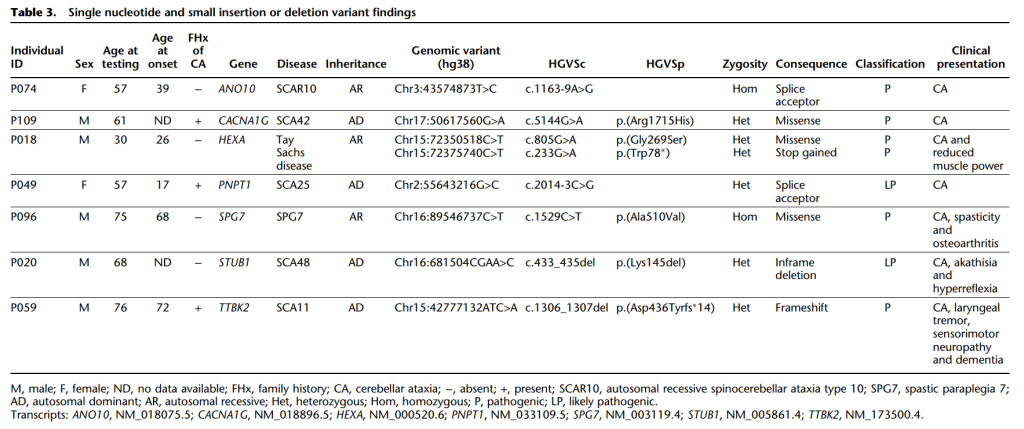
除了RE外,各种其他致病变异也可以引起CA。因此,分析了SR-GS数据中的单核苷酸变异/插入缺失、CNV和线粒体DNA(mtDNA)变异。未发现CNV或mtDNA变异,但通过鉴定ANO10(SCAR10)、CACNA1G(SCA42)、HEXA(Tay-Sachs病)、PNPT1(SCA25)、SPG7(SPG7)、STUB1(SCA48)和TTBK2(SCA11)中的可能致病/致病变异,解决了另外七个患者(表3)。该分析还发现了另外九个个体具有可疑变异,不符合ACMG的LP/P分类标准,因此被归类为意义不明确的变异体。SR-GS的总体诊断率为38%(33个RE,7个非RE),FGF14 RE(SCA27B)的假阳性和假阴性率分别为12%和0%。
长读长测序 (LR-AS) 鉴定重复扩展(RE)
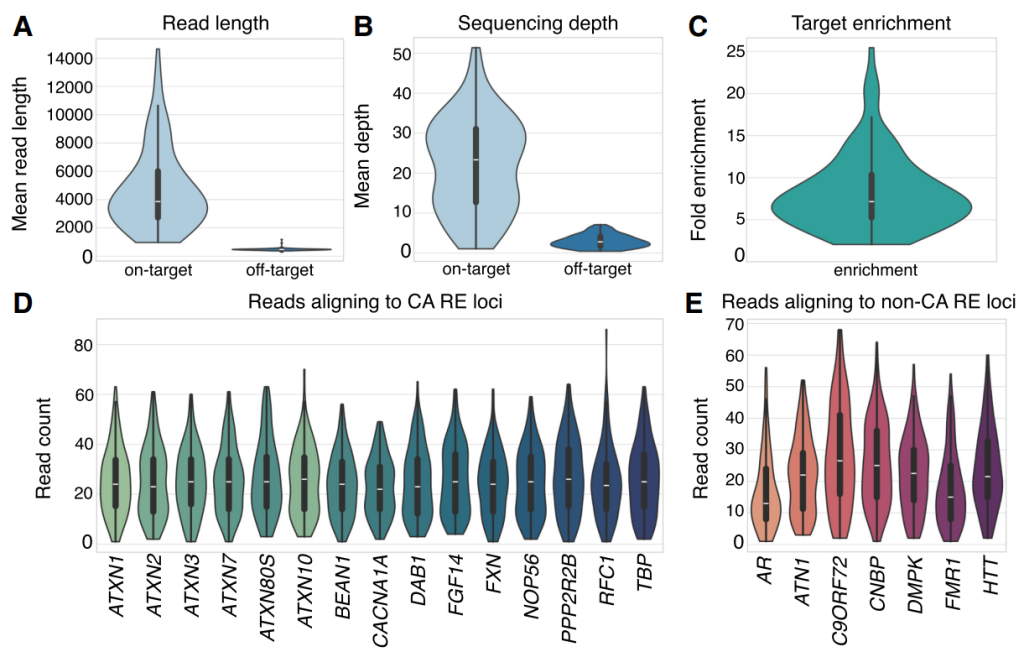
对 334 个基因组区域(111 Mb/约 3.59% 的 GRCh38 [hg38] 参考基因组)进行了长读长适应性采样靶向测序(LR-AS),其中包括67个具有致病性RE的基因以及与CA相关的其他非RE基因,每个样本都使用单个 MinION 测序芯片进行分析。
靶标区域平均读长为 4727 bp,非靶标区域平均读长为 480 bp,靶标区域平均测序深度为 22.1,非靶标区域平均测序深度为 3.0,平均靶标富集倍数为 8.2 倍。
与SR-GS分析一致,我们重点分析了表1中列出的22个RE。在这些22个基因座上,110个个体队列的平均读数范围从16.8个(AR) 到 28.2 个 (C9orf72)。
我们首先测试了LR-AS在识别相应的致病性RE方面的有效性,方法是分析已知具有致病性RE状态的个体
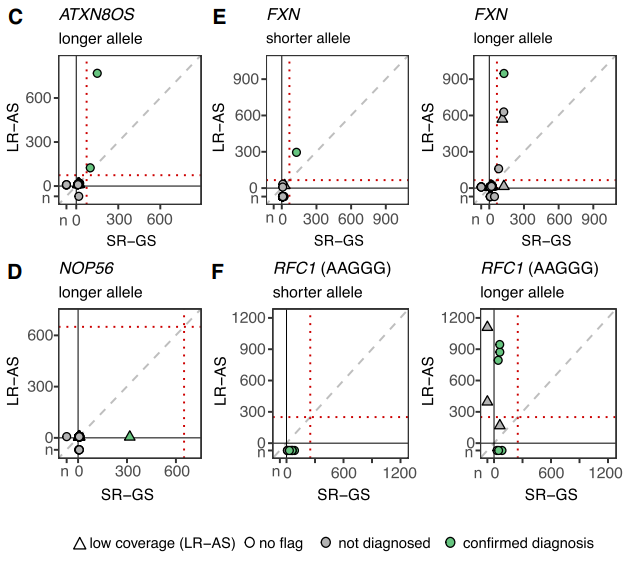
LR-AS识别了29个致病性RE,并为26%的队列实现了基因诊断。这包括ATXN8/ATXN8OS(2个)、NOP56(1个)和FGF14(20个)中的杂合性RE,以及FXN(1个)和RFC1(5个)中的双等位基因RE。这比使用SR-GS识别出的33个致病性RE少4个,所有这些个体均为SCA27B患者,导致差异的主要原因是深度不足。
总而言之,由于其复杂性和变异性,CA的诊断仍然具有挑战性。这项研究比较了 SR-GS 和 LR-AS 在识别 CA 的主要遗传原因——致病性重复扩展 (RE) 的有效性。
SR-GS 的诊断率更高,达到了36%;它受益于成熟的临床应用,能够识别各种遗传变异。它还能支持后续分析,以识别新的RE和CA相关基因。然而,它需要对特定RE进行验证,并且难以检测大型复杂的RE,因此主要用于筛选。
相反,LR-AS 的诊断率达到26%,并且在精确的单倍型解析和靶向分析临床相关区域方面有潜力。尽管需要大量的人工数据分析,并且存在读长和错误率的问题,但 LR-AS 具有发展成为 RE 的独立一线诊断测试的潜力。
参考资料
[1] Rafehi et al., A prospective trial comparing programmable targeted long-read sequencing and short-read genome sequencing for genetic diagnosis of cerebellar ataxia. Genome Res. 2025.
推荐阅读
批次稳定
使用不同批次TargetCap® Core Exome Panel v3.0芯片对NA12878 gDNA进行捕获测序,结果显示,不同批次芯片在不同测序平台上均显示出优异的稳定性,不同位点的相对深度相关性高,批次稳定。