


Genome Research | 长读长测序在疑难罕见疾病诊断上的附加价值
- boke
- 2025-02-18
- 2:28 下午

长读长测序 (LRS) 是一种有前景的技术,有望研究大量未确诊的罕见病,因为它克服了短读测序的许多不足,检测并分析了目前罕见病的标准诊断流程可能遗漏的额外疾病相关变异。
一些可能实现额外诊断的关键领域包括:
(1) 结构变异 (SV) 的检测和分析;
(2) 检测和分析串联重复扩增;
(3) 序列高度相似性区域的测序覆盖;
(4)相位变异;
(5) 利用从头基因组组装,进行基于参考或图基因组的变异检测;
(6) 表观遗传和转录组学分析。
来自超过50项研究的例子支持当前主要的额外诊断产出领域在于 SV 检测和分析、重复扩增评估和相位 (包括或不包括 DNA 甲基化信息)。
一些新研究在未确诊罕见病的队列中应用 LRS 也表明,在常规临床检测结果为阴性后,LRS 可以提高诊断率,并在短读测序结果为阴性后额外提高了 7%–17% 的诊断率。
基于这些改进的诊断率证据,该文讨论了将LRS纳入未确诊罕见病的诊断流程,包括当前的挑战和考虑因素,最终目标是结束无数罕见病患者的漫长诊断之路。
罕见病 (RDs) 是一组多种疾病,在人群中发病率极低,但对全球健康造成巨大负担。RDs 影响每 2500 人中不到1人。
然而,已知有数千种RDs,它们在全球影响范围高达1.5%–6.2%。
大约70%的罕见病在儿童时期发病,高达65%的罕见病与缩短寿命相关,其中约1/4在5岁之前可能危及生命。据估计,约70%的罕见病具有遗传病因。
目前,人类孟德尔遗传数据库报告了超过6400种已知分子基础的表型特征和超过4500个携带致病变异的基因(https://www.omim.org/statistics/geneMap),并且不断发现更多疾病-基因关联。
由于罕见病种类繁多、发病率极低且存在异质性,分子诊断工具至关重要。确定罕见病的分子病因,为患者及其家人提供了获得支持服务、潜在治疗、预后和管理信息、进一步的家庭计划检测,并结束漫长诊断过程的机会。
尽管诊断至关重要,但在标准临床遗传检测后,仍有超过一半的罕见病未被诊断出来。当前的罕见病遗传诊断流程结合了患者的临床表现和潜在的遗传机制,并可能采用各种技术进行分子诊断。
在过去十年中,50-300bp DNA片段的短读长测序 (SRS) 在临床 RD 诊断中日益增多,对靶向区域、蛋白编码外显子(SR-ES)或几乎整个基因组(SR-GS)进行测序。
SRS能以高通量准确评估序列变异(例如单核苷酸变异 [SNVs] 或插入/缺失 <50bp),并能检测某些拷贝数变异和一些结构变异 (SVs,基因组改变>50bp)。
自其广泛应用以来,全基因组SRS,主要为SR-ES,已成为神经发育障碍 (NDDs) 或多发性先天畸形等适应症的有效首选检测方法,诊断率约为30%–35%,这取决于适应症和之前的检测情况。
在进行全基因组短读长测序(SR-GS)后,仍有近2/3的患者未被确诊。尽管使用SR-GS扩展到基因组的蛋白编码区以外,可以更好地评估非编码序列变异,并检测拷贝数变异和结构变异,但目前证据表明其提高诊断率的效果有限,可能仅提升10%左右。
多种因素可能导致在进行全基因组短读长测序后,仍有许多罕见病患者未被确诊,包括对意义不明确变异(VUS)的解读挑战、存在于新型疾病基因中的变异,以及疾病的复杂遗传和/或环境因素。
此外,短读长测序技术固有的局限性也可能是一个重要因素。由于短读长测序的特性,在比对非唯一序列(例如基因组中高度重复或序列相似性高的区域)以及检测和表征结构变异(SV)方面存在困难。
图1A-C展示了短读长测序难以将单倍型拼接在一起或进行从头组装,这给基于读长的变异分型带来了挑战,并限制了复杂基因组重排(CGR)的重建或无参考变异发现方法的应用。
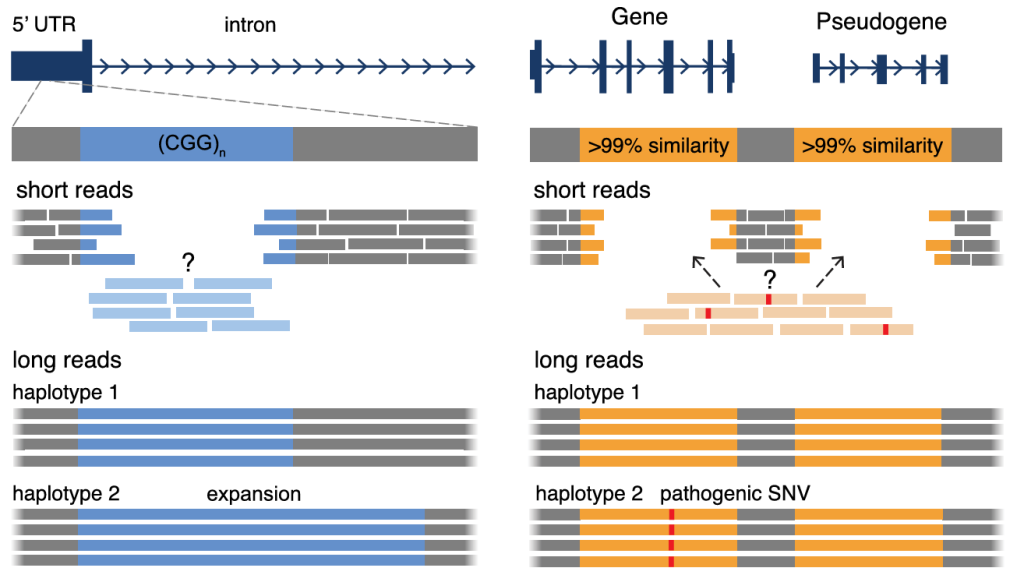
图1. LRS 在未确诊 RDs 中优于 SRS 的效用总结(A-C)。(A) LRS 具有比 SRS 更强的 SV 检测能力,尤其是在 SRS 难以处理的区域,例如重复 DNA(蓝色),重复 DNA 经常介导 SV 的形成。(B) LRS 能够更精准地测序和比对短串联重复序列(蓝色),从而准确检测串联重复扩增。例如,与疾病相关的短串联重复扩增基因包括 FMR1(脆性 X 综合征)、HTT(亨廷顿病)以及与小脑共济失调相关的若干基因(ATXN3、FGF14 等)。(C) LRS 能够更好地对基因组中高序列相似性区域(橙色)进行比对和覆盖,而这些区域对 SRS 来说具有挑战性。这使得能够区分基因与其假基因之间的序列,从而检测这些难以区分的基因的变异。例如,疾病相关基因在高序列相似性区域的例子包括 PKD1(多囊肾病)、IKBKG(X连锁免疫缺陷)和 SMN1(脊髓性萎缩)。
此外,SR-ES中的聚合酶链反应(PCR)扩增步骤可能会引入偏差,并且难以扩增GC含量高的区域(图1D-E)。短读长测序也不允许同时检测天然DNA链的修饰,例如甲基化(图1F)。总而言之,这些局限性可能会导致许多致病变异在进行短读长测序后,未确诊的罕见病患者中无法被发现或解读。
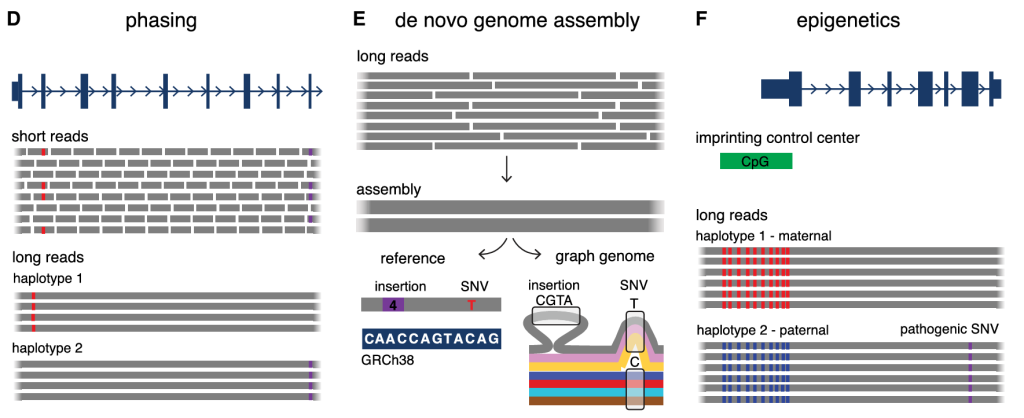
图1. LRS 在未确诊 RDs 中优于 SRS 的效用总结(D-F)。(D) LRS 能够在较长范围内进行单倍型分期,这有助于即使没有父母样本也能确认复合杂合变异(红色、紫色)。(E) 源自 LRS 的长读长可以用于构建高质量且高度连续的从头基因组组装,无需对齐到参考基因组。这些组装体可以与线性参考基因组(左下)进行比较,以检测与参考基因组相比,4 bp 的插入(紫色方框)和 C > T 单核苷酸变异(红色);或者可以与从其他个体获得的从头组装体进行比较(右下,彩色线表示不同个体的组装体),以生成描述个体之间遗传变异的图基因组(只在一个个体中观察到的 4 bp 插入;在群体中更常见的 T/C 单核苷酸变异)。(F) 原始 DNA 链的测序能够同时评估碱基修饰,例如区分 CpG 二核苷酸中的甲基化胞嘧啶(红色)和未甲基化胞嘧啶(蓝色)。这可以结合分期信息用于研究具有亲本来源特异性 DNA 甲基化模式的印记基因座。例如,疾病相关印记基因包括 H19/IGF2(罗素银综合征)、UBE3A(安吉曼综合征)和 PLAGL1(短暂新生儿糖尿病)。
为了克服这些局限性,开发了长读长测序(LRS)技术,能够对天然DNA片段进行全基因组测序,其大小比SR-GS大几个数量级,超过10kb,甚至达到Mb。
这篇综述将回顾LRS在提高未确诊罕见病诊断率方面的现有证据,重点介绍LRS适用的变异类型和情况,以及LRS在未确诊罕见病队列中的应用新证据,并探讨将其纳入罕见病临床诊断路径的考虑因素。
长读测序技术
LRS 技术包括“直接LRS” 或“间接LRS”。在“直接LRS”技术中,直接测序长核酸片段。在“间接LRS”中,例如将长核酸分子的小片段标记,以连接或标记来自同一原始长分子的片段。这些短片段通过 SRS 测序,然后通过生物信息学方法合成重建成原始长片段。
“间接LRS”在单倍型阶段、基因组从头组装和结构变异检测方面优于 SRS。然而,由于测序的基本单位仍然是短读,SRS 的一些局限性可能仍然存在(例如,低复杂度区域的覆盖不足、SV 重建不足、GC 偏差),并且它们在以往的表现不如“直接LRS”。
因此,这可能影响了它们在基因组学研究领域相对于“直接LRS”的应用更为有限。“间接LRS”技术的进一步发展正在进行中,Illumina最近发布的全长测序方法显示出特别有前景,不过在本文中不作为重点讨论。
就“直接LRS”技术而言,两种平台自问世以来一直占据市场较主导地位:PacBio和ONT。
PacBio 高保真 (HiFi) 测序采用的是合成测序法。首先,从大小选定约 15 kb 的文库中提取双链高分子量 DNA,通过连接接头到片段末端进行环化,然后每个环状 DNA 分子都进行多次测序循环,由聚合酶掺入荧光标记的核苷酸,从而实现实时序列测定。这些多次测序的输出(子读段)合并生成共识序列,即 HiFi 读段,其碱基准确性很高,大小通常在 10 kb 至 30 kb 之间。
ONT 使用嵌入电阻膜中的纳米孔,通过电机蛋白将单链 DNA 分子送入纳米孔。当碱基穿过纳米孔时,电流的波动被实时读取,并使用碱基识别算法将其转换为碱基序列。ONT 理论上可以用于测序任何大小的 DNA 片段,这取决于样品输入和文库制备,但通常大于10 kb。目前有用于制备超长读段 (>50 kb) 的文库制备试剂盒(https://store.nanoporetech.com/ultra-long-dna-sequencing-kit-v14.html),研究表明能够生成长达Mb的读段。
尽管这两项技术的早期版本每碱基精度低于常见的 Illumina SRS 平台,但这些精度近年来提高了不少,现在已与 SRS 具有高度竞争力。在约30倍的覆盖深度下,PacBio HiFi LRS 的 SNV 精度 (F1 值) 可达 98.5%–99.9%,indel 精度可达 84.9%–99.4%;ONT LRS 的 SNV 精度可达 98.1%–99.7%,indel 精度可达 69.7%–84.1%,这取决于所用的化学试剂、变异调用器或基准样本。ONT的性能最近通过使用最新R10.4化学试剂的双链测序进一步提高了。
长读长测序带来的额外诊断收益
1.结构变异的检测和解析
基因组结构变异(SVs,基因组改变,大小超过50 bp,包括插入、缺失、重复、倒位、易位和CGRs)在不同个体基因组之间的序列变异中占主要部分,因此在RD诊断中进行全面评估至关重要。
长期以来,人们认识到SVs可能与遗传疾病有关;然而,以前任何单一的临床基因组检测技术都不能准确地评估各种大小和基因组背景下的SVs。
尽管SR-GS有很多SV检测工具,其读取长度明显更长,能够更好地跨越SVs并解析富含SVs的重复序列和非唯一区域,但LRS在检测各种大小的SV方面始终优于SR-GS。
在对比实验中,LRS检测到的SV数量至少是SRS的3到5倍。
到目前为止,检测SR-GS遗漏的SVs提高诊断率的证据主要来自CGRs或对SR-GS具有挑战性的区域/序列的SVs。
例如,PacBio LRS在一位患有复杂神经发育障碍(NDD)的个体中,识别出涉及染色体7和9的多个断点的CGR,其中包含插入易位、倒位和缺失。
例如,涉及转座元件的插入案例也已被报道。这些包括疾病基因SMARCB1和NR5A1的内含子中发现了SINE-VNTR-Alu(SVA)元件的插入,以及LINE-1介导的插入导致CDKL5单外显子重复。
大量证据表明,LRS成功识别了导致疾病的SVs,例如缺失、插入、倒位或更复杂的重排等,这些SV在未确诊的RD患者中被以前的标准护理基因检测遗漏了。由于这些例子中没有进行SR-GS,因此无法证明LRS相对于SR-GS的额外优势。
然而,它们证明了LRS在常规诊断检测后,作为一种检测与疾病相关的结构变异的工具的效用。LRS不仅能识别先前基因组检测中遗漏的结构变异,还能通过精确定位断点和/或揭示结构变异的额外复杂性来阐明已知的结构变异。在8位先前已鉴定出拷贝数变异的个体中,Miller等证明所有重排都能通过靶向ONT LRS识别,并且所有个体都获得了关于拷贝数变异的更多信息,包括精确的断点位置、变异方向以及先前未检测到的拷贝数变异中的额外事件。
这有可能提高诊断准确率,正如在一名先前诊断为平衡易位t(8;18)(q22;q21)的患者身上所体现的那样,ONT LRS在易位位点发现了类似染色体重排的拷贝数变异,涉及19个重排片段,其中包括影响疾病相关基因RAD21和EXT1的缺失,这解释了该患者的症状。
此外,在一项评估短读长基因组测序在神经发育障碍诊断中的研究中,LRS对于完全解析4位患者的复杂结构变异至关重要,从而证明了其相对于短读长基因组测序的优势。
LRS揭示了结构变异的更大复杂性,并/或精确定位断点,从而阐明和证实变异的致病性。这些额外信息突出了其在升级先前未诊断变异并为未确诊神经发育障碍患者提供答案方面的作用。
这在重建和绘制发生在复杂区域(例如片段重复或通常介导复杂结构变异形成的重复元件)的拷贝数变异断点方面可能特别有用。Grochowski等(2024)最近的一项研究证明了这一点,该研究中,LRS有助于解析和精确定位与MECP2重复综合征相关的复杂重复-三联体/反向重复断点,这些断点由附近的反向低拷贝重复介导。
2.串联重复序列的检测和表征
串联重复序列的扩增或减少是另一类关键的SV类型,LRS 可用于诊断和发现未确诊的重复序列疾病 (RD) 的遗传原因。
重复序列是指基因组中 DNA 序列重复多次且相邻的区域,通常分为短串联重复序列 (STRs),重复单元长度为 1-6 bp;或可变数目串联重复序列 (VNTRs),重复单元长度大于7 bp。
据估计,人类基因组中存在超过 170 万个串联重复序列位点,约占基因组的8%。
由于DNA复制错误,串联重复序列高度易变,容易发生扩增或减少。特别是STR扩增,会导致多种重复序列疾病 (RD),特别是成人发作的神经系统疾病,包括几种脊髓小脑性共济失调、亨廷顿病和肌萎缩侧索硬化。
由于其高度重复的性质以及大小通常比单个SRS读长大几个数量级,STR 扩增一直是SRS技术评估的难题。
在临床诊断中,通常需要靶向方法,例如Southern印迹或重复引物 PCR。因此,LRS 有望对STR扩增进行全基因组范围内的准确大小测定和序列组成、表观遗传修饰的表征。
在具有各种已知致病性重复扩增的患者中,对 LRS 进行基准测试的研究始终表明,LRS 可以通过单一综合检测有效地重现分子诊断,方法是识别致病性扩增并提供关于 STRs 的序列组成和单碱基甲基化水平的信息。
这些研究为 LRS 检测致病性扩增提供了重要的原理证明;然而,迄今为止,系统评估LRS检测的扩增大小(特别是大扩增)与标准技术的符合性研究仍然较少。
Stevanovski 等人 (2022) 使用靶向 ONT 技术,与重复引物 PCR 或 Southern 印迹法相比,在 HTT 基因座、FMR1 基因座和 RFC1 基因座上,正常和扩增重复序列长度的R2值分别为 0.996、0.993 和 0.946。
此后,LRS 在发现或进一步表征多种新的 STR 疾病方面发挥了重要作用。这包括最近在ZFX3和THAP11基因中发现的新型扩张,与常染色体显性脊髓小脑共济失调有关;以及HOXD13基因中多组氨酸区段的 27bp 重复,这与综合多指症有关。
此外,LRS也被用于全面表征最近发现的 FGF14 基因内含子(GAA)扩张,该扩张与常染色体显性晚发型脊髓小脑共济失调27B型有关。该 STR 扩张已证实占未确诊脊髓小脑共济失调患者的相当大比例,尤其是在欧洲人后裔中,尤其是在法国加拿大裔中。能够准确测序超过 1000bp 的扩张重复序列证实,只有纯 (GAA)扩张与疾病有关,因为在未受影响个体的大型扩张中发现的富含GA 的基序与所有受影响个体中观察到的扩张 (GAA)基序不同。
由于从 LRS 测序的人群队列中获得的STR基因型数据和分析工具有限,因此从全基因组LRS中发现新的STR 扩张具有挑战性。因此,新的STR扩张发现通常依赖于大型家系,并由连锁研究指导,以识别候选区域,以便进行靶向测序或分析 LRS 数据。随着 STR 基因分型和发现工具的最新改进以及越来越多的群体队列,全基因组分析很快将变得更加可行,并有助于推动更多新的 STR 扩张的发现,这些扩张是未确诊脊髓小脑共济失调的潜在原因。
3.序列高度相似性区域的变异发现
序列高度相似性区域是SRS技术面临的另一个问题区域,因为短序列难以唯一的比对到高度相似的区域,导致这些区域的测序覆盖率很低或缺失。这对于未确诊的遗传性疾病而言非常重要,因为许多与疾病相关的基因都位于这些区域,这些基因可能包含一个或多个假基因(与已知基因高度相似但不会产生功能性蛋白质产物的基因组区域),或基因内部存在多个高度相似的区域。
例如,PKD1、HYDIN、IKBKG 和SMN1,这些基因历来都需要多种靶向分子技术才能全面评估与疾病相关的变异。在 2019 年发表的一项研究中,Ebbert 等人识别出基因体内的36,794个区域,其中包括编码序列中的2855个区域,他们将其称为SRS“暗区”:由于难以充分组装或比对SRS序列,这些区域的测序覆盖率要么很低,要么极低,或者测序读段的比对质量低。他们利用LRS技术的早期版本,证明 PacBio 和ONT LRS 分别显著提高了编码序列中 88% 和 95% 的区域的测序覆盖率。
此外,Wenger 等人发现,在 SRS 暗区中至少有一个外显子的 193 个与医学相关的基因中,有152个(79%)可以使用 PacBio CCS 完全测序。
最近,Kobayashi 等人发现,PacBio HiFi LRS 在他们30名参与者的队列中成功覆盖了全基因组98%的注释SRS暗区,并在一名先前未确诊的免疫系统疾病患者中发现 IKBKG 基因的致病变异,证明了LRS 在这些 SRS 挑战性区域中发现新的诊断的成功应用。
对特定基因的进一步研究也支持了这一点。Borràs 等人的研究发现,对AD多囊肾病患者队列中PKD1和PKD2基因进行靶向 PacBio LRS,鉴定出所有已知的致病变异,具有高敏感性和特异性,并且还鉴定了先前测试中遗漏的额外变异。此外,Fleming 等人的研究表明,使用改进的SRS生物信息学管道,ONT LRS 有助于区分HYDIN 基因与其假基因 HYDIN2的变异,并在原发性纤毛运动障碍患者队列中支持与疾病相关的变异发现。
最近,Chen 等人研发出一种工具Paraphase,该工具能够准确区分 SMN1和其旁系基因SMN2的完整单倍型,从而促进从 PacBio LRS 数据中发现和诊断变异。该工具现在可用于160种长于10 kb的片段重复区域,横跨316个基因,其中包括11个与医学相关的基因。
鉴于人类基因组中超过6000个基因存在 SRS 暗区,这些区域中尚未发现的致病变异可能潜在地导致未确诊的遗传性疾病,而 LRS 有望揭示这些未被发现的变异。
4.相位分析技术改进
将变异体归类到母系或父系染色体,通常是 RD 诊断中的重要步骤。特别是对于具有与隐性疾病相关的基因中复合杂合变异的个体,确定变异体是在顺式(位于同一亲本染色体上)还是反式(位于不同亲本染色体上)至关重要,对于解读变异至关重要。
要直接从SRS读取进行阶段划分,变异体必须足够靠近,以便它们都包含在一个短读或配对末端读中,或者附近有杂合变异体作为参考。或者,如果父母或其他有用的家庭成员的基因型可用,可以通过统计方法确定遗传变异的阶段,利用大型群体数据集的基因型推断相位分型(通常需要基因组测序数据);或者通过更费力的方法,物理分离染色体或选择性扩增一种等位基因,然后再进行测序。相位分型方法在 SRS 数据上应用良好;然而,这些方法对罕见变异的错误率通常较高,在临床 RD 诊断中并不常用。实验室主要依靠家族基因分型或直接读取。
使用显著更长的读取长度,LRS在基于相位分型方面优于SRS,将单倍型块N50(最大单倍型块长度,包含50%杂合位点)至少提高了 10 倍,从约 1 kb 提高到 PacBio 超过 100 kb,ONT 甚至达到Mb。
随着最新测序平台和相位划分工具的出现,基于 LRS 相位分型进一步改进。
使用 PEPPER-Margin-DeepVariant 单倍型感知变异体调用,实现了来自 35× PacBio HiFi 或 25-75× ONT LRS 的 0.24 Mb 或 2-6 Mb N50 的高精度单倍型块。该方法使 35× PacBio HiFi 或 75× ONT LRS 分别能够将 66% 或 93% 的注释基因完全包含在单倍型块中。除了小变异,结合其他变异类型(如 SVs 或 STRs),进一步提高了相位分型。正如一个新工具HiPhase所证明的那样,该工具从 PacBio HiFi 数据中生成了 0.48 Mb 的单倍型块 N50,并完全分离了88%的注释基因。
如果缺乏可供测试的有用家庭成员,或者变异体之间距离过大,无法进行标准临床阶段划分,LRS则有可能为 RD 诊断提供解决方案。
据报道,LRS已成功地对与常染色体隐性 (AR) 疾病相关的基因中的复合杂合变异体进行了相位分型。例如,即使父系DNA无法获得,在两个患有Joubert综合征的同胞中,外显子7中证实了一个LINE-1 插入,其反式与母系遗传的CC2D2A外显子36编码变异体相连,。
LRS提供的易于获得的相位分型信息,也改善了单例数据的分析和解释,尤其是在靶向测序或分析候选基因或区域时。通过靶向 ONT LRS 证明了这一点,他们在9名患有 AR Werner综合征的8例患者中,发现了反式缺失的第二个变异体。
5.基于从头基因组组装和全基因组方法发现变异
LRS相较于SRS,在从头基因组组装和全基因组方法发现变异方面更有效。标准的基于参考的变异检测方法是将测序读段比对到参考基因组,然后用生物信息学工具找出与参考基因组不同的变异。然而,直到最近的CHM13-T2T参考基因组,之前的参考基因组仍然不完整,基因组中存在很多未知序列(缺口),影响了测序读段比对和变异检测。
此外,这些参考基因组仅基于少量基因组的共识,这可能会导致非参考序列识别出现偏差。LRS可以进行从头基因组组装,直接将长读段组装成连续的、解析了单倍型的序列,而无需先比对参考基因组。与标准基于参考的方法相比,基于LRS组装的变异调用可以提高SV和插入缺失的准确性。或者,这些组装体还可以用来发现变异,无需依赖参考基因组,方法是比较多个组装基因组的差异,构建全基因组图谱。这能提高变异检测的准确性,特别是对于SV。
这种方法在RD领域应用还不广泛;然而,最近一项研究表明,从574个儿科RD组装体和94个对照组装体构建的全基因组图谱,提高了SV的可重复性,优于标准的基于参考的方法。它还提高了罕见且可能与疾病相关的SV的优先级,从而在一名之前未诊断的RD患者中发现了KMT2E基因的新型诊断SV。
全基因组方法的局限性包括计算量大,需要针对变异检测开发额外工具。目前,这些方法主要用于提高变异调用准确性和基因分型,尤其是在复杂SV或复杂基因座中。
随着LRS的广泛应用和全基因组参考及分析工具的持续改进,该领域未来可能有助于提高未诊断RD的诊断率。
6.延伸应用
由于LRS技术直接测序未经扩增的DNA,DNA碱基修饰得以保留,并可以通过ONT和PacBio LRS检测,分别基于电信号的特定变化或聚合酶动力学变化。因此,LRS能够以碱基对分辨率评估表观遗传修饰。
这在阐明新发现的RDs的致病机制(例如,在神经元核内包涵体病变相关的NOTCH2NLC中,扩增的(CGG)n等位基因的5-甲基胞嘧啶(5mC)高甲基化)方面非常有用。它还在RDs的分子诊断中具有重要临床意义,其中5mC发生改变。
这包括印记障碍,其中可能通过使用单倍型测序数据和5mC信息检测到印记位点上受干扰的亲本起源特异性DNA甲基化模式,或STR重复扩增疾病,例如脆性X综合征、弗雷德里希共济失调或肌强直性营养不良1型,其中致病性扩增的STRs发生高甲基化。
此外,还在开发全基因组DNA甲基化异常检测方法和工具,以帮助提高未确诊RDs的诊断率。在最近的原理验证研究中,Cheung等人分析了来自152个家系276名个体(均为未确诊儿科RD)的PacBio LRS数据中5mC高甲基化事件,并在DIP2B重复扩增的患者中观察到全面发育迟缓。
因此,LRS结合表观遗传修饰信息和DNA序列,有助于识别未确诊RDs的新候选靶标和诊断。
除了DNA测序之外,LRS更长的读长也扩展到RNA测序(RNA-seq)。RNA-seq可以通过评估疾病相关DNA变异导致的基因表达、剪接和等位基因表达变化来提高RDs的诊断率。然而,之前的研究依赖于SRS,其重建完整mRNA转录本的能力不足,这使得解释剪接变异的确切影响或完全解析新的基因融合变得具有挑战性。相比之下,长读长RNA-seq能对完整转录本异构体进行测序和定量。这推动了人类组织中数千种新型转录本异构体的发现。
它也有助于了解未确诊RDs中剪接变异的影响,例如MFN2中纯合子内含子c.600-31T>C导致5种新型异构体,所有这些异构体都破坏了阅读框并导致无义介导的mRNA降解,以及CLPB中内含子c.1079-23T>A导致一种新型异构体,该异构体具有新的剪接位点,导致CLPB保守的P环插入7个氨基酸。
随着人们越来越关注将RNA-seq作为第二级检测方法来阐明非编码单核苷酸变异(SNV)和结构变异(SV)的转录组改变,在未来,考虑长读长RNA-seq的优势将有助于进一步提高诊断率。
未确诊罕见疾病(RD)队列诊断率的最新研究
目前许多关于长读长测序(LRS)用于提高未确诊罕见疾病(RD)诊断率的研究,主要来自个案报告或已知阳性对照组的验证性研究。这使得准确评估LRS的诊断率提升变得困难。然而,越来越多的研究开始应用LRS于未确诊RD个体队列,部分地展示了LRS的真正潜在诊断率提升。
由于每项研究的指标和之前的基因检测不同,诊断率也存在差异。一些队列研究已将LRS直接应用于标准临床检测之后,使用SRS基因组或SR-ES检测。一项包括34个家系的队列研究中,这些家系患有各种疑似常染色体隐性遗传的罕见疾病(RD),在SR-ES检测后仍未确诊。其中,PacBio LRS数据中纯合性区域分析发现了13个家系的诊断变异,其中8个家系(占总样本的23.5%)无法通过SR-ES检测到。
在更具体的RD亚组中,在SRS检测或SR-ES检测后,LRS的额外诊断率差异很大。这些包括:
1)11个抗凝血酶缺乏症家庭队列中18%的诊断率提升(先前SERPINC1分析结果为阴性);
2)9个肌肉营养不良症家庭队列中44%的诊断率提升;
3)26名结节性硬化症患者队列中50%的诊断率提升;
4)5个遗传性痉挛性小脑共济失调家系队列中最高100%的诊断率提升;
5)最后,Miller 等人在10名患有各种未确诊RD的个体队列中,针对候选基因的靶向ONT LRS测序显示出60%的诊断率提升。
这些个体先前临床检测中已确定为常染色体隐性疾病基因的单一致病变异,或特定疑似常染色体显性或X连锁疾病基因中未发现致病变异。遗憾的是,这些研究无法让我们评估LRS相对于更相似的SRS技术(SR-GS)的诊断率提升。
事实上,一些已识别的诊断变异,例如一些大的缺失和剪接改变的内含子SNV,可能也能通过SR-GS检测到。然而,这些研究仍然支持LRS在典型标准护理检测后的诊断率提高,并可能有助于将来确定LRS在诊断护理途径中的位置。
只有少数报告的研究在SR-GS阴性后将LRS应用于未确诊RD的队列。这些研究对于确定LRS相对于目前可用的SRS技术的额外诊断率提升至关重要。
一些早期研究来自患有未确诊神经发育障碍(NDD)的较小样本量队列(少于10例)。虽然Pauper等人在5名患有未确诊NDD、表现出智力障碍和其他特征的患者中,在PacBio三代测序后未发现任何与疾病相关的候选变异,但Hiatt等人在6名患者中也使用PacBio三代测序检测到2名(33%)患者的可能诊断变异,包括一个新的CGR和一个新的LINE-1介导的插入,两者都影响已知的疾病基因。
值得注意的是,两项研究都只专注于评估新发变异,而新发变异通常是NDD的重要原因。在未确诊RD的异质队列中,进一步的大型研究也支持了相对于SR-GS的递增诊断率提升。
在26个家庭的30名未确诊儿科RD患者中,Sanford Kobayashi等人发现通过单例方法诊断率有所提高,发现了一个SR-GS漏诊的额外诊断,即IKBKG中已知的可能致病的半合子终止缺失突变。
来自“Genomic Answers for Kids project”的另一项大型研究对256名患有各种儿科疾病的受影响参与者进行了测序,其中许多人在SR-GS后仍未确诊。虽然未报告具体的诊断率,但至少有5个案例证明了LRS在诊断中的作用,包括先前未发现的致病重复序列、CGR以及复合杂合变异的相位分析用于支持诊断。
受长读长测序(LRS)通量提高、成本下降以及分析技术改进的驱动,最近对更大未确诊的发育相关疾病(RD)队列进行的全基因组测序,开始揭示LRS相对于短读长测序(SR-GS)的诊断成功率提升。
两项对大约 100 例未确诊的 RD 患者队列的研究表明,LRS 比 SR-GS 的诊断成功率提升在 7% 至 17% 之间。
基于他们 2021 年的研究,Hiatt 等人在96例未确诊的RD患者(表现出神经系统发育障碍、多种先天性异常或疑似先天性肌无力)中应用了PacBio LRS。他们发现了16例患者的新的疾病相关变异或潜在的疾病相关变异,其中7例(占总病例的7.3%)仅通过LRS才能识别。
此外,在对 93 个家系的 232 例未确诊的 RD 患者(表现出神经系统、神经肌肉系统或癫痫症状)的队列研究中,Steyaert 等人利用 PacBio LRS 鉴定了1 例新的诊断(占13%)和4例额外的潜在疾病相关变异(占4.3%)。除了这个未确诊队列,他们还研究了21个家系的小队列,这些家庭患有罕见且临床可识别的未确诊综合征(如 Aicardi 综合征、Hallermann-Streiff 综合征、Gomez-Lopez-Hernandez 综合征、眼-耳-椎体谱系疾病),但未在任何这些综合征中识别出受影响个体之间共享的候选基因或基因区域。
总之,这些队列研究结果令人鼓舞,表明 LRS 相对于 SRS 技术的诊断成功率有适度但显著提高,最终为无法通过其他方法诊断的家庭提供了诊断。值得注意的是,这些研究主要关注影响已知疾病相关基因座、新的体细胞变异或大型外显子重叠变异的变异。随着分析方法和对照队列的不断改进,随着研究界更好地利用 LRS 在未确诊的 RD 中的全部潜力,这些诊断成功率可能会进一步提高。
长读测序在RD诊断流程中的应用探讨
尽管研究表明长读测序(LRS)在未确诊的RD中具有应用价值,并有望提高诊断准确率,但从样本制备到分析的几个因素仍然限制了这项新技术的广泛应用于RD诊断流程。
首先,当前的文库构建步骤需要相对大量的优质、高分子量DNA才能获得全基因组LRS的长读长。这需要使用能够保持这些大DNA分子完整性的生物样本和DNA提取方法,这在样本量少或难以获取血液等侵入性样本时可能会受限。
第三,LRS的历史成本较高且通量较低,这也导致了控制数据集的获取较为有限,用于变异等位基因频率的注释。鉴于LRS在检测SV和覆盖具有挑战性的SRS区域方面具有更高的灵敏度,来自同样经过LRS测序的人群队列的等位基因频率数据库对于RD中的罕见变异分析至关重要。不幸的是,目前公开可用的LRS基因组数量与SRS数据集(例如gnomAD)相比仍然较少。令人鼓舞的是,最近的一些努力已开始取得进展。这包括CoLoRSdb(https://colorsdb.org/),这是一个汇编数据资源,包含来自多个队列的超过1400个PacBio LRS基因组,包括“Human Pangenome Reference Consortium”、“Human Genome Structural Variant Consortium”以及RD队列,例如“Genomic Answers for Kids project”。
此外,最近发布了来自1000个基因组计划的1019个样本的数据集,这些样本由ONT LRS以中等覆盖率(16.9x)测序,适合SV分析,以及来自1000个基因组计划ONT测序联盟的第一个100个样本,这些样本的测序深度至少为30x。
随着更多人类基因组通过LRS测序,这些等位基因频率数据库将继续增长和改进。事实上,“All of Us”倡议最近进行了一项可行性研究,建立了一种大规模使用PacBio进行LRS的方法,以在人群水平上准确发现小变异和SV,为其计划的人群规模LRS工作做准备。
通过注册访问其研究工作台(https://www.researchallofus.org/ ),现在可以获得来自前1000名参与者的数据。
最后,限制LRS整合到RD诊断流程中的第四个关键因素是数据分析和处理所需的基础设施。虽然存在许多用于LRS数据分析的工具,以支持初始碱基调用、比对、组装、分相、变异调用等,但生物信息学和分析流程的最佳实践和标准尚未统一。这些对于确保临床实验室间和实验室内的结果一致性是必要的。还需要考虑处理和存储这些全基因组数据所需的基础设施。所有这些限制都是积极发展的领域,并且在过去几年中得到了显著改善,这使得将LRS应用于临床流程变得越来越可行。
简化临床基因检测流程,提升RD患者获得全面基因检测的便捷性,最终通过比现有临床标准更快、更广泛地识别分子诊断,从而缩短诊断时间。如果考虑多项样本采集、处理、培训、分析、报告以及级联检测中每个环节的额外成本,使用LRS作为单一的一级检测,在某些情况下可能会更有效率和更经济。但这尚未得到充分证明,因此,评估LRS作为一级检测相较于标准临床流程的诊断效率、临床价值和经济效益至关重要。
将LRS作为一级检测应用于需要快速、全面诊断的病例或可由单一LRS检测替代多步骤复杂临床检测的病例,有助于证明其效用(图2)。
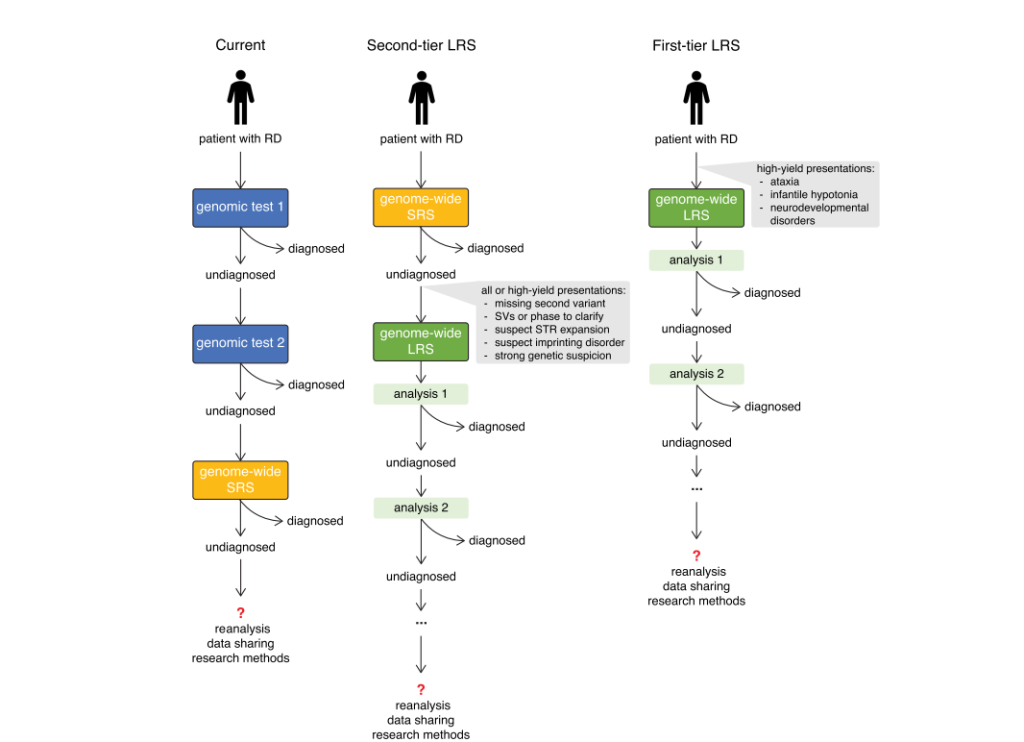
图 2. 假设将LRS纳入未确诊RD治疗路径。(左)目前RD患者的诊断路径,患者可能经历漫长的诊断过程,根据需要接受多次不同的基因组检测,包括全基因组SRS。(中) 建议将 LRS 作为全基因组 SRS 检测结果未明确诊断的二线检测。研究表明,在全基因组 SRS 检测后纳入 LRS 可提高诊断准确率;因此,该路径有可能减少未确诊 RD 患者的数量。(右)未来将 LRS纳入一线检测的建议,这主要适用于那些 LRS 对这些 RD 来说可能更经济的情况。这简化了逐步的诊断流程,但允许对 LRS 数据进行连续分析(例如,编码变异、重复序列、结构变异、甲基化修饰等)。
例如,将LRS作为危重症患者快速诊断的一级检测,或作为评估所有已知致病性重复扩张的单一检测,适用于出现共济失调的患者。此外,对于肌张力低下症状的婴儿,一级LRS检测非常有吸引力,因为当前标准可能包括多种检测,例如核型分析、微阵列分析,以及根据临床怀疑对多种基因进行靶向评估,包括脊髓性肌萎缩症的SMN1和SMN2基因、肌营养不良症的DMPK基因STR扩张以及/或Prader-Willi综合征的15q11.2区域的甲基化和拷贝数检测(图2)。迄今为止,研究表明,LRS作为全面评估与共济失调相关的STR的一级检测是可行的,并在儿科重症监护环境中提供超快速诊断。随着LRS成本的持续下降、通量的增加和分析的成熟,我们预计该技术将更快地融入诊断护理途径。
结论与未来展望
LRS是应对这一挑战的关键技术,可整合到未确诊RDs的护理流程中。LRS已证明能够识别SRS的挑战性变异,解决已知的疑难变异,例如,提供表观遗传学等额外支持信息,以提高未确诊RDs的诊断准确率。
进一步改进通量、降低成本、增加可用的控制/队列数据以进行等位基因频率注释,并标准化分析方法,将支持将该技术纳入临床诊断实验室。
鉴于LRS领域取得的显著进展,本文认为,现在是更广泛地应用LRS来解决剩余未确诊RDs问题的时候了。相信这项技术将帮助我们更接近为所有RDs实现准确及时诊断的目标。
目前,伯科生物长读长靶向捕获技术已经与众多国内外研究机构开展合作,广泛应用于多个临床与转化领域,包括遗传病诊断、辅助生殖以及肿瘤研究等方向。
让辅助生殖技术(PGT-M)惠及更多患者| 伯科靶向长读长Gene Panel助力动态突变精准检测
6 基因靶向长读长检测,进一步提升听力障碍患者分子诊断阳性率
MLPA+Sanger+SRS+LRS | 伯科长读长靶向捕获Gene Panel助力常染色体显性多囊肾的复杂遗传病因解析
[1] Del Gobbo and Kym M. Boycott. The additional diagnostic yield of long-read sequencing in undiagnosed rare diseases. Giulia F.Genome Res. 2025