


MRD检测之广度换深度
2021-11-30
检测0.01% VAF的ctDNA并不容易,Ellen Heitzer等用泊松分布模拟了含有6000个基因组拷贝的样本在VAFs为0.01%、0.1%和0.5%时(~20ng input DNA)的重复抽样情况(图1)。当VAF为0.5%时,99.5%的样本含有10个或更多的突变拷贝。相比之下,在VAF为0.01%时,绝大多数样本将不包含任何突变拷贝。
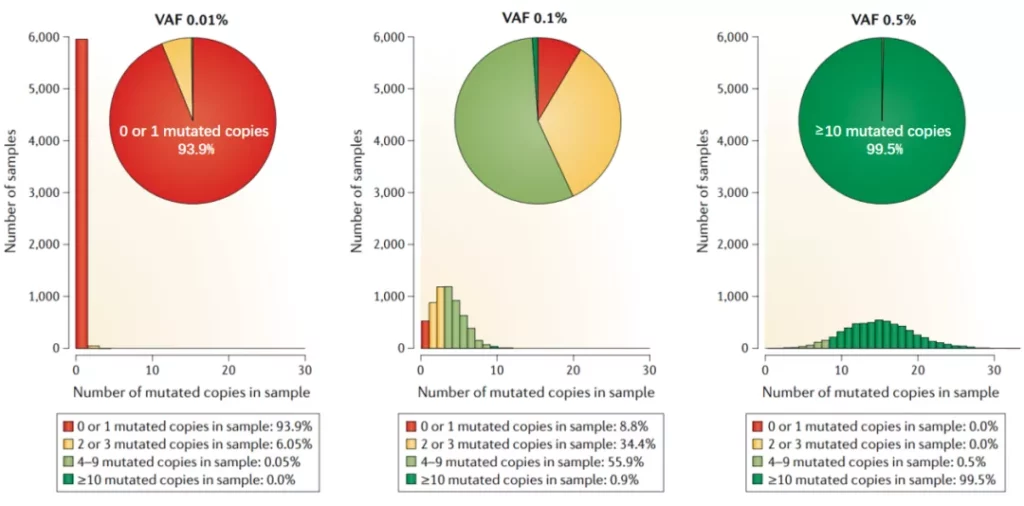
图1.对6000个拷贝的样本进行不同VAFs重复取样后突变拷贝的泊松分布[1]
如果能够获得充足的cfDNA,达到足够的测序深度,也是能够改善超低频检测的困境。但事实上,血浆cfDNA是有限的,比如10mL的采血管,实际分离出的血浆只有~4mL[1],据报道,癌症患者的cfDNA浓度在10ng/mL左右,而健康人群为~6ng/mL[2]。按照健康人群估算,采血20mL能够获得的cfDNA为24-48ng,约7000-14000个基因组拷贝。按照实验损耗50%计算,真正能够用于检测的只有3500-7000个拷贝,如果要检测0.01% VAF频率的ctDNA基本上是捉襟见肘。
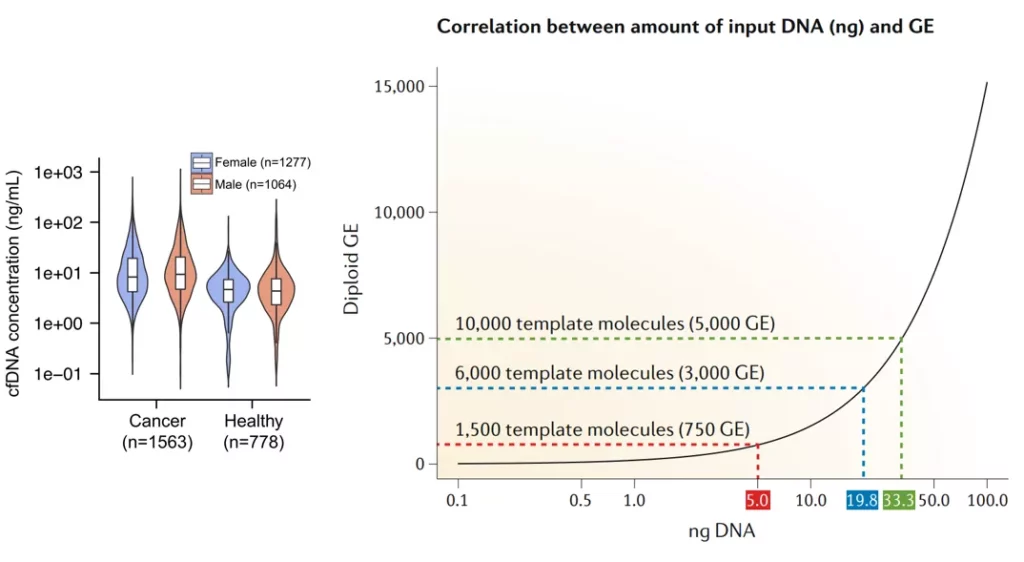
图2. cfDNA浓度与基因组拷贝[1,2]
那么,如何提高MRD检出?Asaf Zviran等利用cfDNA样本突变片段抽样概率模型预测ctDNA检出概率(至少检测到一个突变拷贝)与tumor fractions(TFs)以及基因组拷贝(unique genomic equivalents)之间的关系,结果表明通过增加检测位点(SNV)的数量,可以有效地克服投入量限制。如图3a所示,待检测SNV数量为1时,检出概率随着TFs的降低而迅速降低;SNV数量为10个,TFs为10-4时,在5000个拷贝中的检出概率超过80% [3] 。另一项报道的模拟数据也表明同时监测多个突变可以增加ctDNA的检出概率,Ymke Van Der Pol等利用二项式模拟1mL血浆cfDNA(10ng[3000 copies] cfDNA除去损耗后剩余的1500个拷贝)中检测到至少一个ctDNA的概率(图3b),结果表明增加突变数量在理论上可以提高低VAF下的ctDNA的检出[2]。即便低起始量cfDNA限制了ctDNA的检测灵敏度,也可以通过增加检测突变数量加以克服。
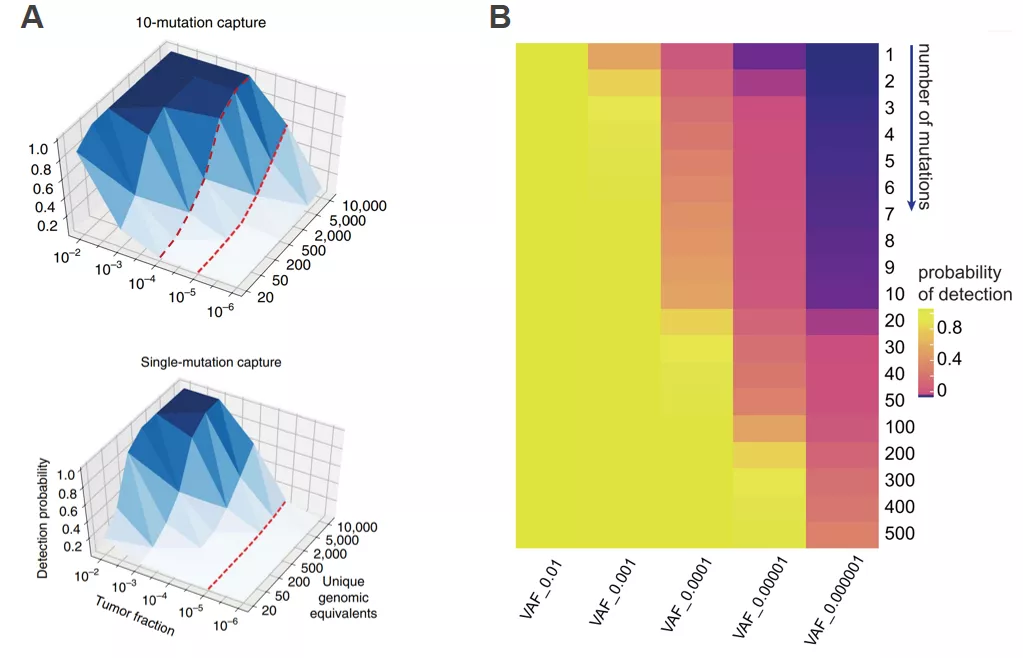
图3. 增加跟踪突变数量可提高突变检出概率[2,3]。a. 在不同肿瘤占比和基因组拷贝情况下,检测不同数量突变与检出概率之间的关系;b. 模拟计算1mL血浆cfDNA在损耗后(1500 copies),不同VAF情况下,待检突变数量与检出率之间的关系(至少检测到一个突变拷贝)。
因此,更多的突变位点是MRD检出率的间接保障,但先决条件是肿瘤携带足够的变异供我们筛选。根据先前FM的TMB大队列研究,不同癌种的TMB中位值为3.6 mutations/Mb,这一数字因癌症类型有较大差异,从0.8-45.2 mutations/Mb不等(图4)。在其他研究中,黑色素瘤和肺癌平均大于8.9个突变/Mb,而胶质瘤、乳腺癌、胰腺癌或前列腺癌平均小于2.2个突变/Mb[5,6,7]。需要注意的是,在癌症的早期阶段TMB通常更小,这意味着可被检测的突变只会更少[8,9]。筛选出的突变再经过生物学和技术过滤,比如克隆性和引物或探针设计,真正能用于后续监测的突变位点数量并不乐观。
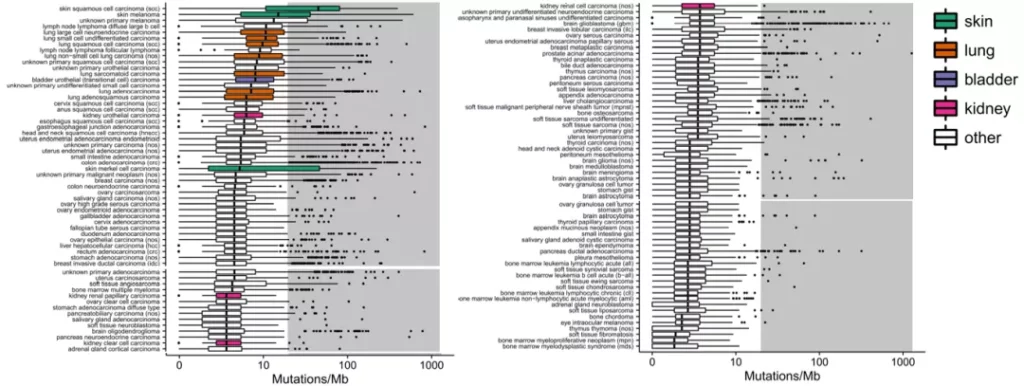
图4.不同癌种TMB分布[12]
因此,尽可能扩大基因组筛选范围就显得非常重要,WES通常覆盖30-60Mb左右的基因组Exon区间,使用WES进行肿瘤组织测序能够为后续的MRD检测提供更加充足的突变位点候选,为MRD检测灵敏度保驾护航[10,11]。
2020年,Nature杂志报道了Jacob J. Chabon等人的相关研究,使用靶向捕获技术,对于85例I-III期非小细胞肺癌患者,使用355Kb固定Panel只能在cfDNA中跟踪4个突变(中位值),整体ctDNA检出率为49%(38/85);与上文的理论估算趋势一致,ctDNA检出率和跟踪突变数量正相关。而对17名患者采用WES测序进行突变筛选,随后采用个性化定制Panel进行cfDNA监测(SNV数量中位值68,Panel Size 212 – 487 Kb),ctDNA检出率提升至59%(10/17),中位VAF更是低至0.002%。值得一提的是,研究中有个非常有意思的地方,个性化Panel设计中,研究人员将5-7名患者的突变位点集合成一个“个性化”Panel并进行检测,该方案可以简化检测实验,有借鉴意义。
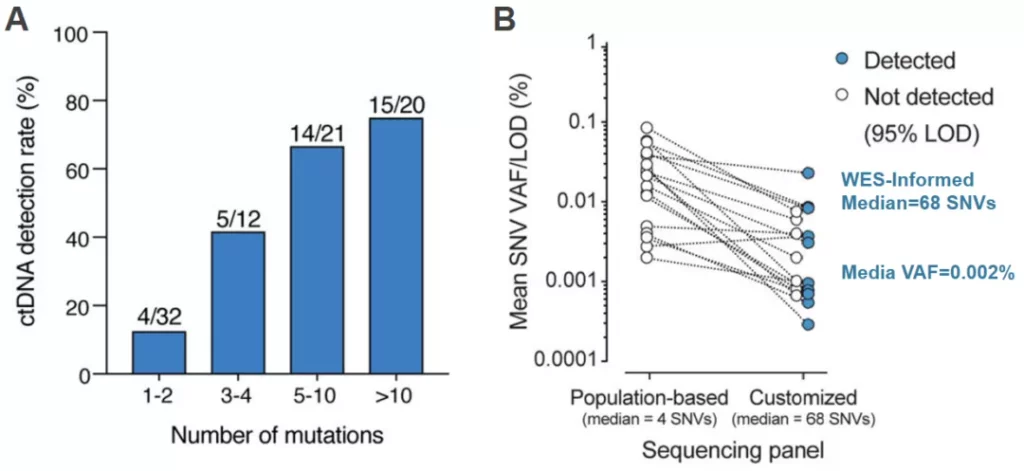
图5. ctDNA检出率与跟踪突变数量正相关,WES+个性化Panel能够追踪更多的突变位点,提高ctDNA检出[13]
所谓“巧妇难为无米之炊”,由于生物学方面的限制,血浆cfDNA的含量以及肿瘤组织的突变数量都是有限的;那么,在成本和周期都可控的情况下,搜索出尽可能多的突变位点,采用针对更多突变位点的个性化Panel进行跟踪,能更好的做到“物尽其用”。
参考文献
[1] Heitzer E , Haque I S , Roberts C , et al. Current and future perspectives of liquid biopsies in genomics-driven oncology[J]. Nature Reviews Genetics, 2018.
[2] Pol Y , Mouliere F . Toward the Early Detection of Cancer by Decoding the Epigenetic and Environmental Fingerprints of Cell-Free DNA[J]. Cancer Cell, 2019, 36(4):350-368.
[3] Zviran A , Schulman R C , Shah M , et al. Genome-wide cell-free DNA mutational integration enables ultra-sensitive cancer monitoring[J]. Nature medicine, 2020, 26(7):1-11.
[4] Pol Y , Mouliere F . Toward the Early Detection of Cancer by Decoding the Epigenetic and Environmental Fingerprints of Cell-Free DNA[J]. Cancer Cell, 2019, 36(4):350-368.
[5] Brennan C W , Verhaak R , Mckenna A , et al. The Somatic Genomic Landscape of Glioblastoma[J]. Cell, 2013.
[6] Rotin D . Comprehensive molecular profiling of lung adenocarcinoma[J]. Nature, 2014.
[7] Mutational heterogeneity in cancer and the search for new cancer-associated genes[J]. Nature.
[8] Garcia-Murillas I , Schiavon G , Weigelt B , et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer[J]. Science Translational Medicine, 2015.
[9] Direct detection of early-stage cancers using circulating tumor DNA[J]. Science Translational Medicine, 2017, 9(403):eaan2415.
[10] Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution[J]. Nature.
[11] Gremel G , Lee R J , Girotti M R , et al. Distinct subclonal tumour responses to therapy revealed by circulating cell-free DNA[J]. Annals of Oncology, 2016, 27(10):1959-1965.
[12] Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden[J]. Genome Medicine, 2017, 9(1):1-14.
[13] Chabon JJ,Hamilton EG,Kurtz DM,et al. Integrating genomic features for non-invasive early lung cancer detection[J]. Nature,2020,580(7802).


