


遗传病基因变异全外显子组测序技术规范化 应用专家共识
- boke
- 2025-02-25
- 4:05 下午

遗传病是指基因变异或染色体数目和结构异常所导致的疾病,已成为当前全球公共卫生领域的重大挑战。已明确的遗传病超过7000种,其种类繁多、表现形式及病因机制各异[1]。遗传病的基因检测在产前筛查、出生缺陷预防、新生儿筛查、遗传病防治等方面均具有重要的价值[2-4]。
外显子是基因组中负责编码蛋白质的区域。尽管仅占基因组全长的1% ~ 2%,却包含近85%的已知致病变异[5]。与全基因组测序相比,全外显子组测序(wholeexomesequencing,WES)具有成本低、效率高的优势,因此被广泛用于遗传病的检测。然而,与传统的检测相比,高通量测序技术操作复杂,数据分析和变异解读人员能力参差不齐,检测方法多为实验室自建,实验室标准化和规范化不足,造成了结果的不确定性。相较于其他临床检验项目,遗传病基因检测具有很大的特殊性,临床医师在疾病诊断和干预方面对结果的依赖程度更高,检测结果可能直接影响后续的诊断和预后,因此对检测的准确率和质量要求更为严格。为保障 WES技术在遗传病检测领域的规范应用,在参考国内外指南、标准、规范的基础上,本共识组列出了WES检测的适应症,包括产前诊断的适应症,并在实验室设置、操作流程、数据处理、结果解读、质控等方面提出了规范和建议。
WES检测的适应症
WES检测的适应症包括:
(1)未确诊的遗传性疾病:综合患者的临床症状、影像学及常规实验室检查结果、家族史等高度怀疑为遗传性疾病,但传统的诊断方法(如染色体核型分析、染色体拷贝数分析等)未能提供明确的遗传学诊断结果;
(2)罕见病和综合征:罕见的遗传性疾病或综合征,如不明原因的智力落后和(或)发育迟缓、非已知综合征的多发畸形等;
(3)复杂性遗传病:临床诊断明确、但表现出复杂遗传模式的疾病或存在较强遗传异质性的疾病(如眼科疾病、骨骼发育异常等);
(4)产前诊断:胎儿影像学检查发现明显异常,但常规染色体核型分析或拷贝数分析无法提供明确诊断者。此外,对于一些临床诊断明确的遗传病患者,或具有反复流产或异常生育史的夫妇,WES可以帮助识别潜在的遗传因素,为再生育提供指导。
2
实验室设置
2.1 设施及环境要求
实验室应遵照《医疗机构临床基因扩增管理办法》的相关规定开展工作,并符合生物安全法的有关规定。测序区域应具备满足测序仪等设备正常运行所必需的实验室洁净度、温湿度、防震和光照等条件。实验室应依据所用设备和实验过程的要求,满足对环境温湿度的要求并进行监控。各实验区应安放专用的仪器设备。不同区域的仪器设备、一次性耗材等物品不应混用。
2.2 人员要求
检测人员应具备检验资质并经过省级及以上具有资质的机构的培训并取得临床基因扩增检验技术上岗证。
从事生物信息学分析的人员应熟练掌握人类遗传学知识,具备对高通量测序数据进行分析和基因变异解读的能力。变异解读等关键岗位的人员应掌握足够的医学知识,熟悉相关疾病的临床表型,并定期学习国内外专业学会发布的人类基因组变异解读的相关指南和标准。实验室应定期对检测人员进行培训。
新上岗的人员应完成岗前培训、考核和能力评估,在合格后方能独立开展检测。应妥善保存培训记录、考核资料和评估记录。
2.3 仪器设备、试剂及关键耗材要求
实验室应建立仪器设备使用、维护、检定校准的程序文件,并按照文件严格执行。影响检测结果的关键仪器设备应定期进行检定/校准,并有合格的校准报告。
所有试剂均应严格按照要求妥善保存并在有效期内使用。关键耗材(离心管、吸头等)应不含抑制物。应选用带滤芯的吸头。不同批号的试剂组分不能混用。
3
样品要求
在采集样品时应选择正确的采集/储存容器,并采集足够量的样品。样品采集完成后,应在容器外标明样品的编号、种类、患者姓名以及采样日期。应按照相关规定对样品进行包装和运输。样品采集后应尽快转运。若需要长途运输,则应采用干冰等制冷方式进行保存。
样本在采集后应尽快进行检测,并选择适宜的温度进行保存。能够在24h内检测的样品可置于4℃保存;24h内无法检测的样品则应置于–70℃或以下进行保存(若无–70℃的保存条件,则应置于–20 ℃冰箱内暂时保存),并且避免反复冻融。
在检测完成后,应根据样本的类型及相关规定来确定保存的时间和条件。检测后的样品应按照生物安全法的相关要求及时处理。
4
检测要求
4.1 核酸提取
应针对不同的样本类型选用适当的基因组 DNA提取试剂,按照试剂盒说明书进行操 作。提取后的DNA 应进行质量检测。总量应不低于200ng,A260/A280> 1.8。必要时应进行电泳检测,电泳条带应无明显降解。
4.2 文库制备
应按照选用的文库构建试剂盒的说明书进行操作。纯化好的文库在稀释后应进行定量分析。合格标准为:浓度应与相应测序平台相匹配,总量不低于 5ng,片段长度为200 ~ 500bp。应根据测序接头唯一的原则制定文库混合方案,在进行等比例混合后,使用荧光定量等方法测定文库的浓度。
4.3 高通量测序
应按照测序试剂和仪器的说明书进行操作。测序数据的质控要求为:单个样本有效测序 通量为 8~10Gb,碱基测序质量 Q30 ≥ 85%,GC含量为40% ~ 55%。
4.4 生物信息学分析
测序产生的原始数据的分析流程包括:fastq处理、比对、bam 处理、变异检测(产生vcf文件)、变异注释、致病性分析及结果判读、质控等。
分析软件:各种生物信息学分析软件正式在使用前均需进行分析性能验证,证明所用的软件及其各项参数设置均满足临床报告的要求。在改变或更换软件时,需重新进行性能验证。所有分析流程使用的软件和参考序列均需记录相关的版本号,以确保准确分析,以便后续回溯。
参考序列及比对数据库:应使用当前公认有效的人类基因组参考序列(如美国国立生物技术信息中心NCBI收录)和注释最全面的数据库进行分析。
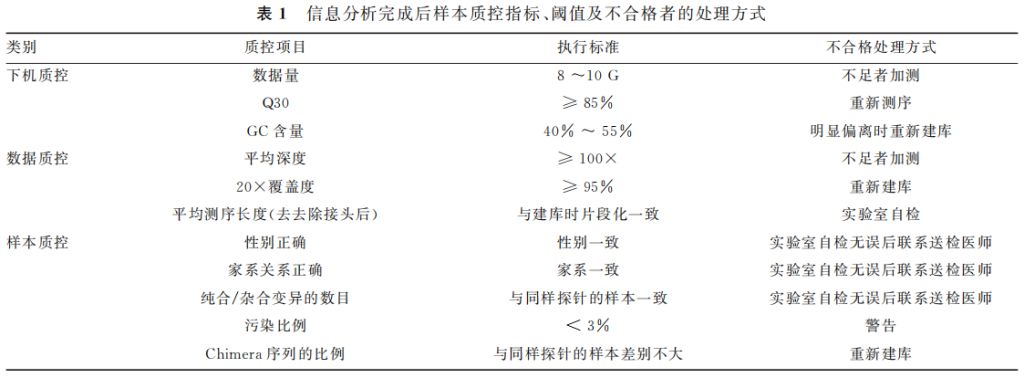
数据分析的最低要求:测序数据在去除重复后建议平均深度≥100×,20×的覆盖度必须≥95%,测序片段平均长度200 ~ 500bp。外显子区域至少包含目标基因的一致性编码序 列 (consensuscoding sequence,CCDS)± 10bp。对结果的一致性进行初筛:分析结果性别正确,同一家族成员之间亲缘关系正确,污染比例< 3%,纯合/杂合变异和嵌合体序列比例与同批次样本一致。样本数据的质控指标及不合格的建议处理方式见表1。
5
变异的解读及分级
按照基因变异与疾病的关系确定候选变异的致病性等级,将其分为致病性(pathogenic,P)、可能致病性(likely pathogenic,LP)、意义未明 (variant of uncertain significance,VUS)、可能良性 (likely benign,LB)和良性(benign,B)。
参考美国医学遗传学与基因组学学会(American Collegeof MedicalGeneticsandGenomics,ACMG)变异解读指南以及美国临床基因组资源中心(ClinicalGenomeResource,ClinGen)序列变异解读工作组和英国临床基因组科学学会 (AssociationforClinicalGenomicScience,ACGS)等对该指南的细化规则[6-20],收集变异位点致病性的支持证据,综合进行变异的致病性判断。综合受检者的临床症状及家系遗传模式对结果进行分析,确定与疾病相关的变异报告位点。
5.1 判定基因与疾病的关系
基因与疾病的关联可分为6个等级:明确证据(Definitive)、强证据(Strong)、中等证据(Moderate)、有限证据(Limited)、无证据(NoEvidence)、证据冲突(Refuted/Disputed)。基因-疾病关联的评级细则可参照 ClinGenGeneDiseaseValidityCurationSOP(https://www.clinicalgenome.org/site/assets/files/9232/gene_curation_sop_version_10_1_docx.pdf)。当基因与疾病的关系判定为有限证据时,致病性的评级最高为意义不明(VUS)。在产前诊断时,通常仅报告强 度 为 Definitive/ Strong 的 基 因,报 告 强 度 为Moderate的基因时应谨慎。
5.2 特殊的基因变异评级
针对一些特殊的基因及疾病,需要采用 ClinGenVCEPs特定标准进行评级,具体参照 https://www.clinicalgenome.org/affiliation/vcep/# ep _ table _heading。
5.3 转录本的选择
转录本应参照 HGMD 数据库转录本和MANE转录本 (https://www.ncbi.nlm.nih.gov/refseq/MANE/),基因名称需遵循国际人类基因组组织(TheHumanGenomeOrganisation,HUGO)推荐的基因命名法(http://www.genenames.org),变异名称需遵循人类基因 组变异学会 (HumanGenomeVariationSociety,5H.G4VS各)推条荐评的级命依名据方的法细(氨则基酸和蛋白质水平)。
5.4 各条评论依据的细则
5.4.1 类 别 1———等 位 基 因 频 率:BA1,BS1,PM2_Supporting
BA1:适用于在任一人群数据库中等位基因频率(allelefrequency,AF)> 5% 的变异,且至少观察到2000个等位基因,同时无特殊针对基因或变异的BA1建议。
在gnomAD数据库中可以获取特定人群亚群中的等位基因频率、来源、等位基因变异总数以及所有人群中的等位基因总数。
原始变异和变异热点内的变异频率可以升高。由于疾病患病率、外显率和基因异质性的不同,BA1的阈值在不同的基因/疾病中可能存在差异。对于某种疾病,BA1所反映的是特定基因的贡献,而 BS1则代表最大可信变异的最大等位基因频率。BA1和BS1 的 阈 值 可 以 使 用 在 线 应 用 程 序 进 行 估 算(https://www.cardiodb.org/allelefrequencyapp/)。
对那些在特定种族群体中常见的变异,等位基因频率 的 阈 值 存 在 一 些 例 外 的 情 况 (https://www.clinicalgenome.org/site/assets/files/3460/ba1_exception_list_07_30_2018.pdf)。
BS1:适用于等位基因频率大于疾病发病率的变异。
一般来说,如果该变异在正常人群中比在疾病中出现的频率更高(或至少1种gnomAD 中的亚群),可认为是较强的良性影响证据。ACMG/AMP 指南建议变异频率的阈值需要综合考虑疾病的发病率和外显率。等 位 基 因 频 率 的 估 算 可 以 使 用 在 线 软 件(https://www.cardiodb.org/alleleFrequencyapp/)。
BS1 的 最 小 等 位 基 因 频 率 (minor allelefrequency,MAF)的阈值因疾病而异。在某些情况下,例如听力缺失或 RAS综合征,BS1可以作为独立证据使用,将变异评级定为“可能良性”(表2)。
BS1_StandAlone(BS1_SA):对具有高外显率的常染色体显性遗传病,可以使用BS1作为独立证据(BS1_SA)将变异归类为“可能良性”(https://www.acgs.uk.com/quality/best-practice-guidelines/#VariantGuidelines)。
PM2_Supporting:适 用 于 gnomAD、ESP 数 据、1000G 或 ExAC等数据库正常人群中未发现的变异位点或极低频变异位点。
对于某些在特定种族群体中常见的变异,应考虑等位基因频率阈值存在 的 特 殊 情 况 (https://www.clinicalgenome.org/site/assets/files/3460/ba1_exception_list_07_30_2018.pdf)。
5.4.2类别 2———变异的类 型 及 位 置:PVS1,PP2,BP1,PP3,BP4,PS1,PM5,BP7,PM4,BP3
PVS1:当某种疾病的致病机制为功能丧失(lossoffunction,LOF)时,对无功能变异(无义变异、移码变异、经典±1或2的剪接变异,起始密码子变异、单个或多个外显子缺失、基因内重复(≥1个外显子))评级时使用。证据的强度水平应根据变异的类型和特征进行调整,具体如下:
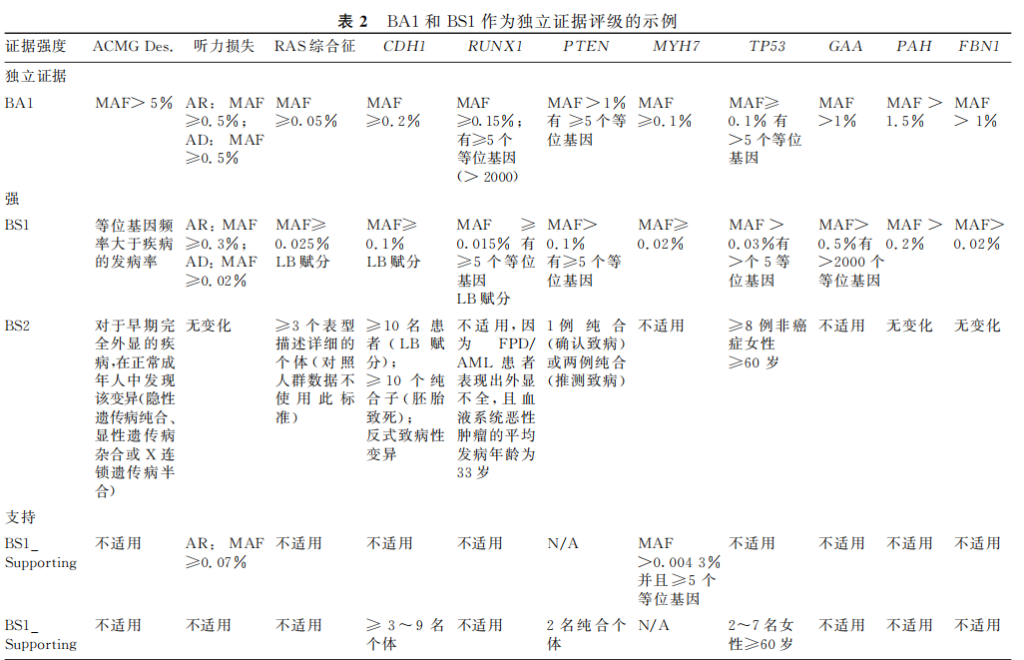
(1)起始密码子:
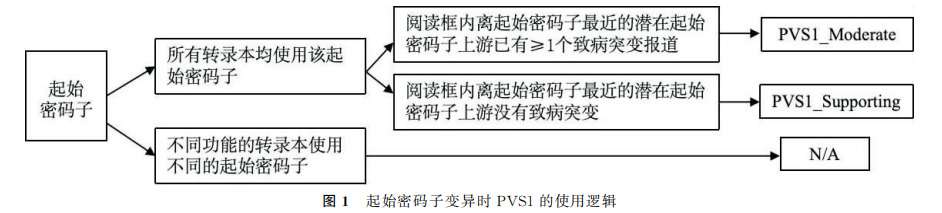
(2)无义变异和移码变异:
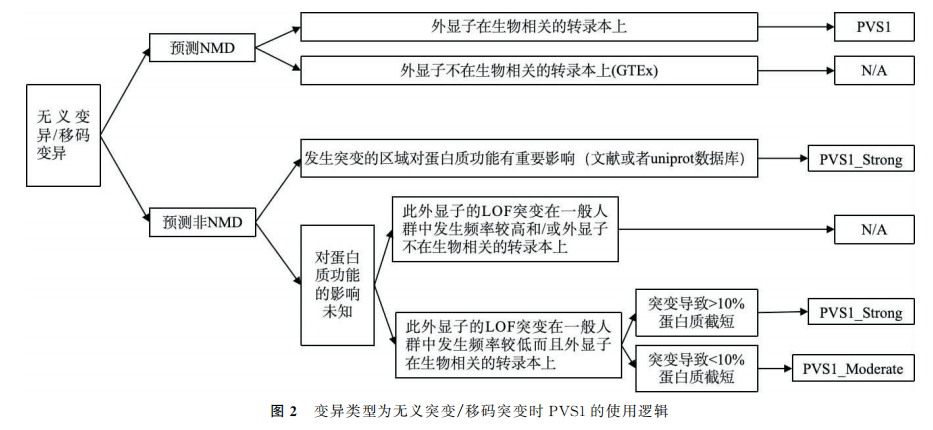
(3)单个外显子至整个基因缺失:

(4)基因内部外显子的重复:
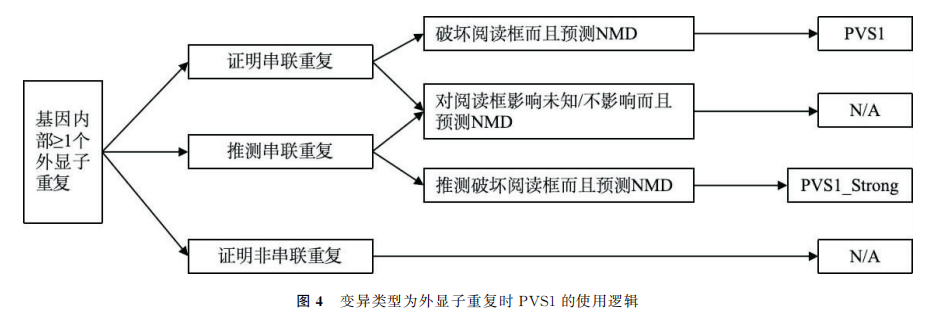
在使用PVS1时应注意:(1)PVS1和PM4不能同时使用,PVS1和 PP3不能同时使用;(2)在使用 PVS1前必须 明 确 基 因 与 疾 病 的 联 系,并 确 定 功 能 丧 失(LOF)为其致病机制。致病机制不是 LOF的基因(如GRAP、MYH7 等)不能使用 PVS1;(3)解读基因3’端的 LOF变异时需谨慎;(4)应谨慎对待可能导致外显子跳跃的剪接变异,因其可能产生仍保留功能的蛋白;(5)在存在多个转录本时,应结合与疾病受累组织的相关性综合考虑。
PM4:适用于非重复区的框内插入/缺失或终止密码子丧失造成蛋白质长度变化的变异。
在应用 PM4时,需明确变异所改变的蛋白质区域是否在功能上重要且保守。建议对涉及单个氨基酸的框内缺失或插入(in-framedeletionsorinsertions)使用 PM4_Supporting。除非有 证 据 证 明,则 可 以 使 用Moderate。例如文献报道在同一氨基酸上与疾病有关的错义变异。终止密码丧失导致3′-UTR 出现框内终止密码子时,也应使用 PM4。
BP3:适用于功能未知的重复区域内未造成基因阅读框改变的缺失/插入变异。
PP2:应谨慎使用。PP2 适用于特定基因内的错义变异:基因的错义变异是导致某种疾病的原因,并且该基因中良性变异所占的比例很小。一般来说,如果某个基因的错义变异的Z 值≥3.09(gnomAD查询),则表示对错义变化不耐受的程度增加,可以使用 PP2。Z 值越高,对变异的不耐受程度就越高。重要的是应考虑变异区域的容忍度,而不仅是整个基因 (参见DECIPHER数据库)。
BP1:已知疾病的致病原因为某个基因的截短变异,对该基因中发现的错义变异可使用 BP1。
PP3:适用于使用多种统计方法预测会对基因或基因产物造成有害影响的变异。统计算法包括保守性预测、进化预测、剪接位点影响(SpliceAI>0.2)等。由于许多生物信息学算法均使用相同或非常相似的输入进行预测,因此单个算法不应被视为独立标准。PP3在任何情况下对每个变异只能使用一次。ClinGen推荐使用REVEL来代替单个的预测工具。REVEL是12种不同生物信息学预测工具的集合。其评分范围为0 ~ 1,其中1预示强致病性。REVEL 分值≥0.644可以使用PP3(https://clinicalgenome.org/docs/variantcurationstandard-operating-procedure-version-3/)。
BP4:适用于多种统计学方法预测对基因或基因产物无影响的变异,这些算法包括保守性预测、进化预测、剪接位点影响等。若 REVEL分值< 0.290,可以使用 BP4(https://clinicalgenome.org/docs/variantcurationstandard-operating-procedure-version-3/)。
PS1:适用于与已确定的致病性变异有相同氨基酸改变的变异。即不同碱基变异为同一个氨基酸时候使用,且已报道的变异的致病性必须经过评级后证实为致病者(P/LP)才可以使用。对一些特殊的基因,请参照其特定标准进行评定,并且须确认该变异并非通过影响剪接而改变基因的功能。PS1也适用于已被确定为 P/LP的起始密码子变异,因其具有相同的分子结果,即起始密码子丢失(p.Met1?)。
PM5:适用于同一氨基酸位置上新的错义变异,即该变异既往未见报道,但在同一位点造成的另一种氨基酸变异已被确认为致病性(P/LP)。如果新的错义变异所涉及的氨基酸上至少已有两个其他的 P/LP错义变异,则可以使用 PM5_Strong。同时须确认该变异并 非 通 过 影 响 剪 接 而 改 变 基 因 的 功 能。在 使 用PM5前,应使用 Grantham 分值来比较这两个变异。新的变异应具有与已确定的致病变异相同或更高的Grantham 评分。若无法应用 Grantham 评分标准(新变异的评分低于先前分类的 P/LP变异),则可以使用PM5_Supporting。
BP7:适用于同义变异且预测不影响剪接。BP7也适用于内含子或非编码区的变异,但不包括以下区域:内含子内的-3~-1核苷酸之间的常规共识剪接位点;内含子内的十1~ 十6 核苷酸之间;在外显子的第1 个核苷酸或最后2个核苷酸上。
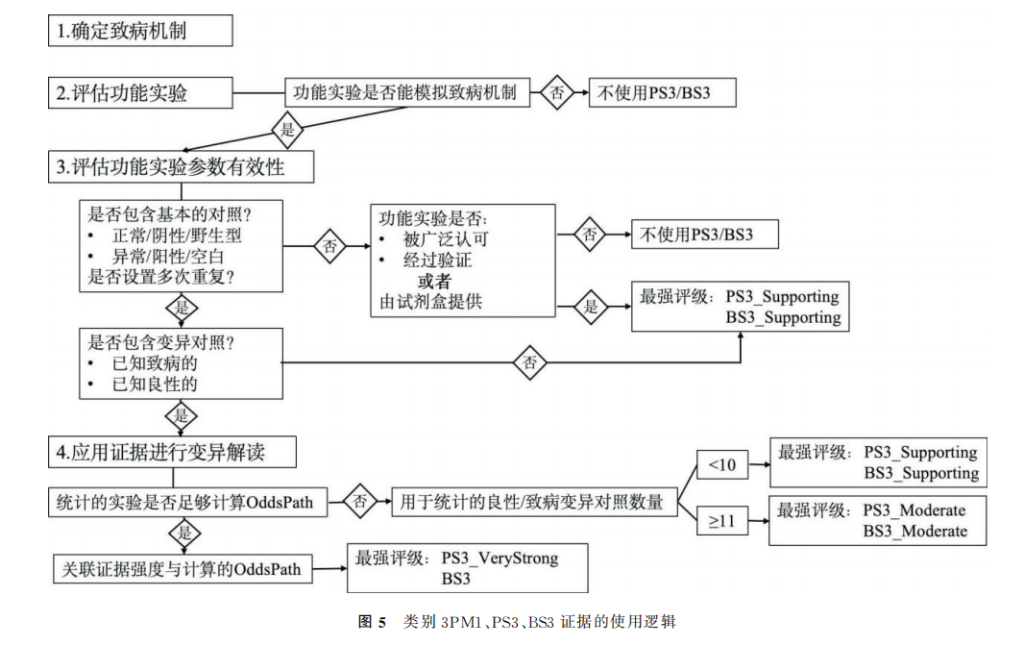
5.4.3 类别3———功能性数据:PM1,PS3,BS3
PM1:适用于位于热点变异区域和/或无良性变异的关键功能区的错义变异;
PS3:适用于体内和体外功能实验已明确会导致基因功能受损的变异;
BS3:适用于体内和体外功能实验明确不会影响蛋白功能或剪接的变异。
5.4.4 类 别 4———患 者 和 家 族 成 员 共 分 离 的 数 据:
PS4,BS2,PM3,BP2,PS2,PM6,PP1,BS4,PP4,BP5
PS4:适用于在患病群体中的频率显著高于对照群体变异。
BS2:适用于分析发育早期完全外显的疾病时在健康成人中发现的变异(如分析隐性遗传病时发现的纯合子、分析显性遗传病时发现的杂合子以及分析 X连锁遗传病时发现的半合子)。
PM3:适用于分析隐性遗传病时检测到患者中与P/LP变异呈反式位的变异,且该变异需满足 PM2_Supporting。
根据所讨论的两个变异的相位和另一个等位基因变异的分类,每个先证者都会获得一个分值(表3)。
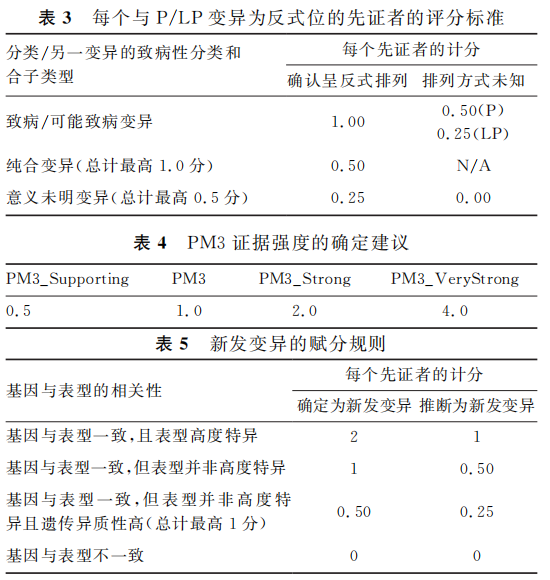
所有先证者事件的分值总和将被用于确定证据强度的级别(表4)。
应注意等位基因的频率:若两个等位基因的频率均满足 PM2阈值的要求,则可将先证者的评分作为 PM3的证据,以避免由于高频率变异恰好与 P/LP变异呈反式排列而导致PM3的误用。若变异在一般人群中的等位基因频率较高,则应使用病例-对照证据(case-controlevidence,PS4),而不使用PM3病例证据。
BP2的:适用于分析显性遗传病时与致病变异呈反式排列的变异以及任何遗传模式疾病中与致病性变异呈顺式排列的变异。
PS2:适用于确认过父母血缘关系的新发变异。
PM6:适用于未确认过父母血缘关系的新发变异。
PP1:适用于在家系中符合共分离规则的变异。
BS4:适用于在家系中不符合共分离规则的变异。
对于常染色体显性遗传病:
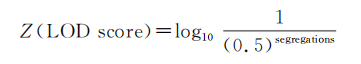
根据家系中患病家系成员数量判断LOD值(每个得分0.3),多个家系的 LOD 值可相加。2 ~ 3个为Supporting,4个为 Moderate,5个及以上为Strong。
对于常染色体隐性遗传病:
应根据患病家系成员和未患病家系成员的数量进行判断,具体见表9。
患病的家系成员数 = 患者数-1,且为复合杂合或纯合变异未患病的家系成员数:通常指未患病的同胞数(有遗传与先证者相同的2个变异的风险、但两个位点均为野生型或单位点为杂合变异者,父母不算在内)。
PP4:适用于变异携带者的表型或家族史高度符合某种 单 基 因 遗 传 病 者。可 使 用 等 级:PP4,PP4_Moderate。PP4不能用于非特异性表型,例如发育迟缓、智力障碍、自闭症、癫痫或心肌病等。一些专家组已将 PP4 归 纳 为 其 他 证 据 类 型 (PS4、PS2/PM6、PP1)、而非单独使用 的 证 据。若 有 非 遗 传 分 析 的 结果,例如代谢紊乱先天性异常的生化特征(苯丙酮尿症中血苯丙氨酸水平> 120μmol/L 等)或特征性表型(例如 Hunter综合征的皮肤卵石样特征),也可以使用 PP4。
BP5:适用于在已有另一明确致病原因的病例中发现的变异。
5.5 位点的致病性判定
汇总所收集到的人群数据、文献病例数据、特定变异类型分析及各类软件计算预测等证据进行变异致病性判定,具体参照表10。

6
报告位点的确认
6.1 应根据变异对应的疾病与受检者临床症状的吻合度、致病性解读结果对变异进行优先级排序,具体规则如下:
第一梯度:主要症状吻合+致病性包含P/LP+遗传模式相符;
第二梯度:主要症状吻合 + 致病性仅为 VUS+遗传模式相 符;主 要 症 状 吻 合 且 为 医 师 重 点 怀 疑 的基因/低遗传异质性疾病变异(无论遗传模式是否相符);
第三梯度:次要症状吻合+致病性仅为 VUS+遗传模式相符;
第四梯度:主要/次要症状吻合 + 致病性仅为VUS+遗传模式不符。
6.2 应根据优先级确定拟报告的变异。
报告结果分为主要检测结果、次要检测结果、意外发现3类:
主要检测结果:可解释受检者主要/部分表型的P/LP变异、遗传模式相符的可能解释受检者表型的意义不明变异、复核医师诊断的疾病/基因的意义不明变异。即属于第一和第二梯度变异。
次要检测结果:其他与表型可能相符的变异,第三、四梯度的变异。
次要发现:只报告 ACMGSFv3.2所列的81个基因,有文献报道、遗传模式相符且致病性为 P/LP的变异。默认报告,若患者/家属选择不报告,则需签订相关的知情同意书。
6.3VUS的报告逻辑
临床意义不明变异(VUS)通常不进行报告。但在某些情况下,存在较高的支持证据,并可能根据相关证据来重新分类为(可能)致病性时应考虑报告。根据ACGS指南,VUS变异致病性的强弱可根据相关证据进行进一步的分类和打分(表11),超过3分者可考虑报告。
7
报告撰写
7.1 确定报告的变异
根据Sanger测序验证的结果和变异致病性的解读结果确定报告的变异。
7.2 变异描述
应参照 HGVS 推荐的标准基因变异命名法进行描述,具体参见https://hgvs-nomenclature.org。
7.3 报告撰写
撰写变异解读结果以及所采用的证据、疾病描述等。

8
检测结果的质控
8.1 质量管理体系
实验室应编写操作规程及相关的记录表单,内容涵盖检测的所有环节(包括湿试验和干实验)。在实验过程中应严格执行标准操作规程,不能擅自修改。
8.2 性能确认
在正式开展 WES检测项目前,实验室应明确其预期的临床用途并进行方法学的性能确认,相关参数包括但不限于精密度、准确度、可报告范围、检出限、抗干扰能力,必要时还应进行临床验证。
8.3 室内质控
建议每批次检测至少包括1份阳性质控品(可使用标准品)和1份阴性质控品。室内质控品应与待测样品同时参与全程检测,包括核酸提取和扩增的全过程。在判断结果时,阳性质控品应检出,阴性质控品结果应为阴性。若失控,不可发出报告,应及时分析原因,必要时重新进行检测。
8.4 室间质量评价
实验室应每年参加省级及以上临床检验中心组织的室间质量评价。
8.5 检验结果的可比性
若实验室使用两套及以上的检测系统(仪器/试剂),则应有比对数据表明检测结果的一致性。若为手工操作,则需进行不同操作人员之间的比对。应每年进行至少1次比对,样本数量不少于5份,包括不同的变异类型(如单核苷酸变异、插入/缺失、拷贝数变异等)和阴性样本,结果应全部符合。
伯科全外芯片 – Core Exome Panel v3.0
TargetCap@ Core Exome Panel v3.0基于伯科高品质DNA探针合成技术开发,全流程国产制造,由~40万条探针组成,以GRCh38/hg38人类参考基因组设计,参考Refseq、CCDS、ClinVar等数据库,覆盖19,524个基因,目标区域为33.9Mb。
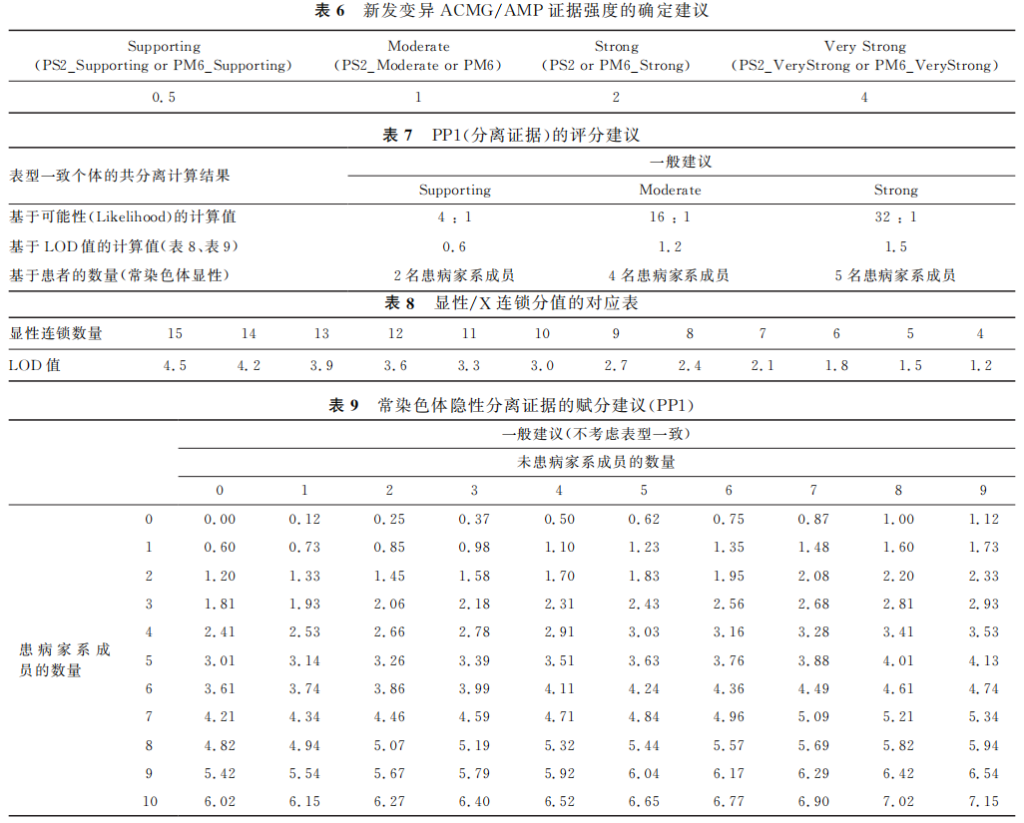
捕获性能比较
伯科全外芯片v3.0性能优异与国外友商同类型产品v2相当,中靶率、覆盖率、覆盖均一性等参数均达到国际领先水平。
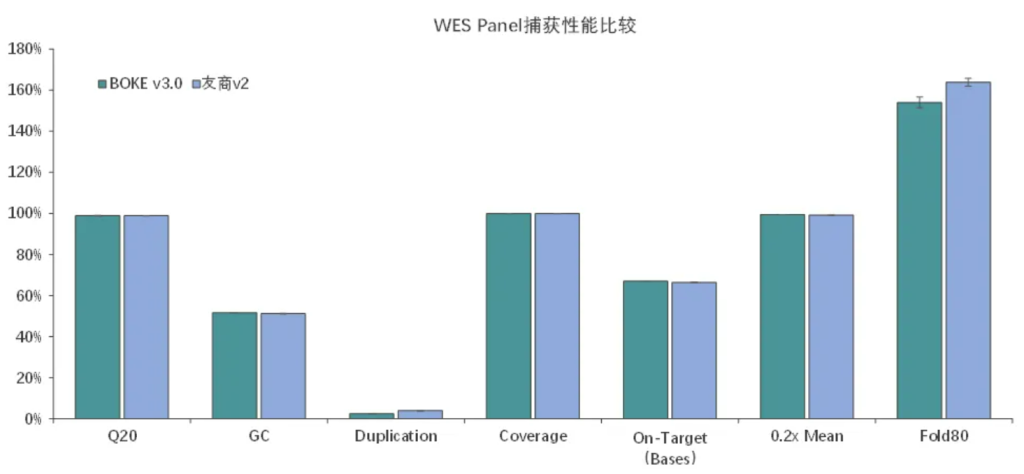
适配高通量流程平台
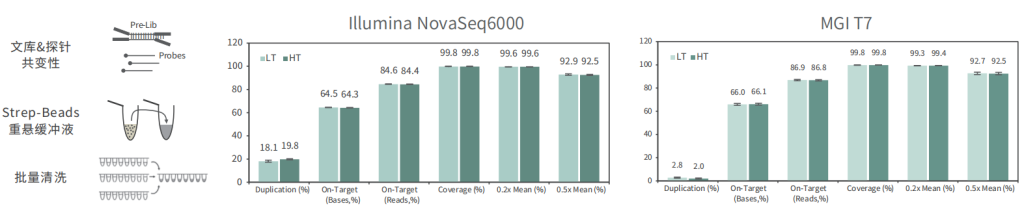
批次稳定
使用不同批次TargetCap® Core Exome Panel v3.0芯片对NA12878 gDNA进行捕获测序,结果显示,不同批次芯片在不同测序平台上均显示出优异的稳定性,不同位点的相对深度相关性高,批次稳定。
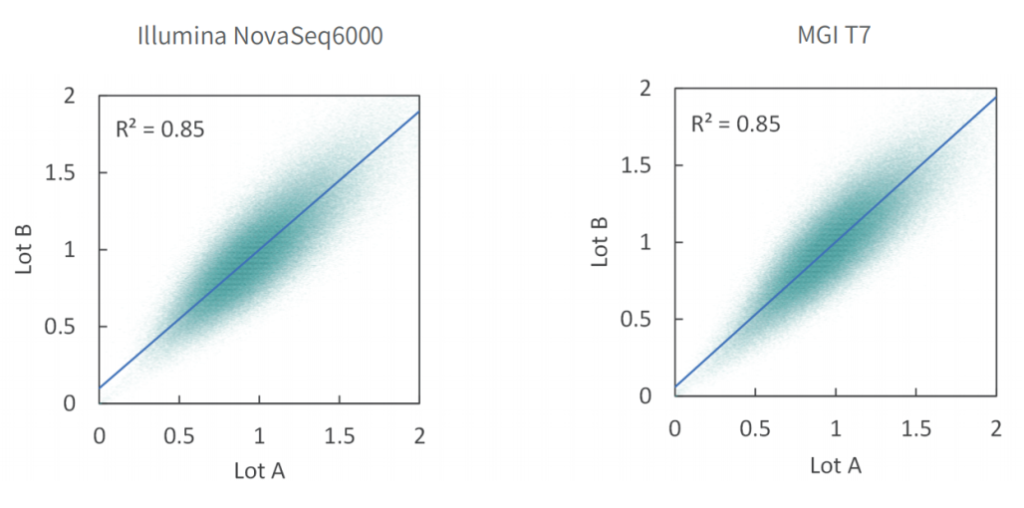
变异检测准确
单核苷酸变异(SNV)和插入缺失 (INDEL)是基因组变异的常见形式,也是引起人类疾病的重要原因。
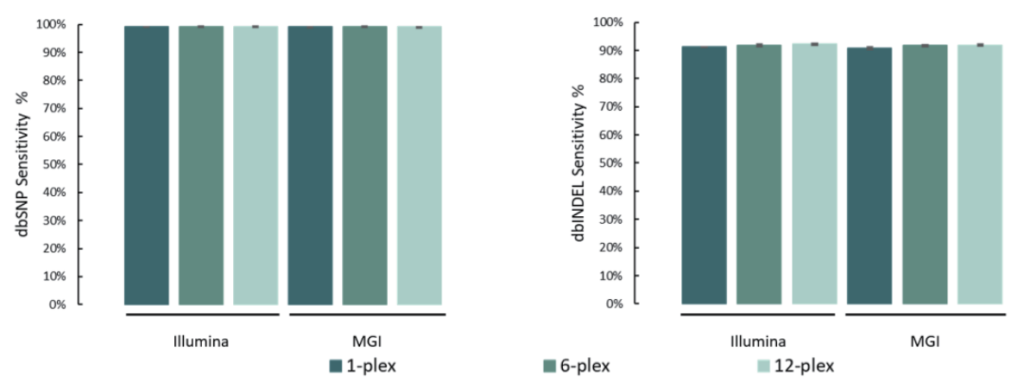
选取NA12878标准品,与预期SNV和INDEL变异进行比较。结果表明,在MGI与Illumina测序平台,SNP灵敏度为99.1%,INDEL灵敏度为91.6%。
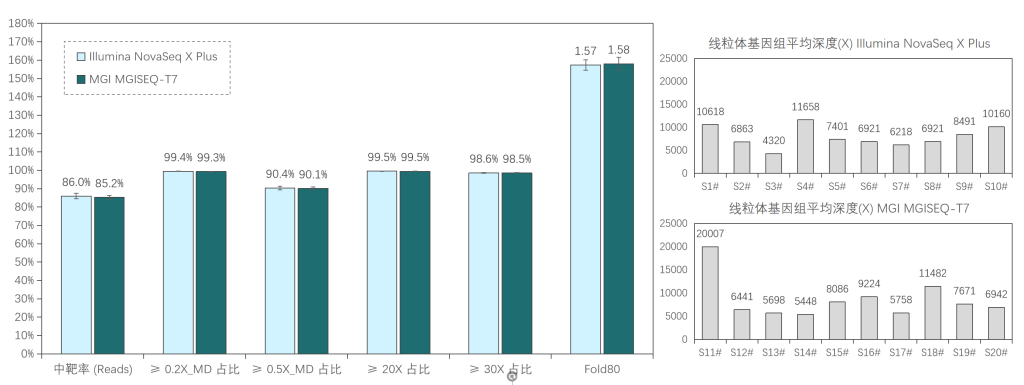
添加线粒体模块临床样本表现
20例全血样本(S1#-S20#),采用1-4 Plex方式使用伯科Core Exome Panel v3.0添加线粒体模块进行过夜杂交捕获;其中,S1#-S10#在Illumina NovaSeq X Plus平台测序, S11#-S20#在MGI MGISEQ-T7平台测序,均采用150PE模式测序。得到测序数据后,抽取8Gb数据进行生信分析。
两种测序平台的数据表现相近,平均深度分别为111x/115x (Illumina/MGI),中靶率优异均> 85%,覆盖均一性极佳(0.2X_MD≥99.3%);仅使用8Gb数据,高达98.5%的捕获区域达到了30X以上,99.5%的捕获区域达到20X以上,为临床样本检测提供了可靠的捕获数据。
参考文献:
[1]遗传病基因变异全外显子组测序技术规范化应用专家共识.中华医学遗传学杂志2025年1月第42卷第1期