


Cell综述 | 遗传性心肌病的研究与治疗进展
- boke
- 2025-07-01
- 5:10 下午
心肌病的患病率高,病因复杂,表现多样,预后不良,且与遗传密切相关,这些特点使其成为精准医学理念指导下,心血管疾病诊、治、防、控的重要领域。
近年来,随着影像与分子遗传技术的进步,心肌病在精准诊断与个体化治疗等方面不断取得更新和突破,国内外心肌病管理指南与共识陆续更新,心肌病的诊疗迎来了一个“复兴时代”。

对疾病分子基础的遗传学发现为更精确的诊断和治疗奠定了基础,并揭示了导致临床特征差异的机制。这种更深入的理解推动了心肌病新疗法的开发:包括疾病特异性的、基于机制的对抗病理生理学的或旨在阻止疾病进展并恢复心脏功能的紧急基因疗法。这些发现让我们对心脏生物学有了更深入的了解,并标志着心肌病患者的新时代。跟随Cell的综述,让我们一起了解遗传性心肌病的最新研究进展。

引言
心脏肌病,作为原发性心脏肌肉疾病,传统上根据解剖形态学和相关的临床特征进行诊断。三种主要表型占主导地位:肥厚性心肌病 (HCM),其特征是心脏肌肉增厚、收缩力增强和舒张功能障碍;扩张性心肌病 (DCM),表现为左心室扩大和收缩功能障碍;以及心律失常性心肌病 (ACM),以与心脏结构重塑程度不成比例的严重心律失常为特征。
这种分类方法实用,并推动了改善发病率和死亡率的疗法的发展。然而,这种分类方法存在局限性,因为它依赖于相对低分辨率的临床特征,这些特征通常并非疾病类型的独有特征。它也未能充分反映个体患者的复杂性,或区分对治疗有反应或无反应的患者亚组。揭示导致这些差异的关键机制将能够实现更准确的诊断、改进临床预后的预测以及更精确的靶向治疗。
过去三十年间,在确定许多心脏肌病的分子遗传基础方面取得了显著进展。尽管在病因学和表型上差异很大,但约 65% 的 HCM 患者在编码心肌肌节蛋白(心肌细胞的收缩单位)的基因中存在致病变异。肌节蛋白变异多见于家族性疾病患者和儿童期 HCM 患者,而成人发作的 HCM 较少表现为孟德尔遗传。DCM 和 ACM 的遗传基础更为复杂,并且由于临床表现的异质性、年龄依赖性和不完全外显率,发现工作面临挑战。
与心肌肌节蛋白、细胞骨架和细胞间连接相关的基因参与疾病发生,但仅占所有病例的一半以下。然而,建立 HCM、DCM 和 ACM 的遗传病因具有重要的临床意义,因为遗传学研究日益指导临床管理。我们回顾了目前对遗传性心脏肌病的理解,并讨论了将临床护理发展为更具机制性方法的机会和挑战。遗传诊断目前如何影响管理?基于机制的药物(包括基因疗法)的未来发展方向是什么?实现这些目标的障碍是什么?
肥厚型心肌病(HCM)
疾病定义
传统上,肥厚性心肌病 (HCM) 的临床诊断依赖于左心室肥厚 (LVH),其原因并非由压力过载(例如高血压或主动脉瓣狭窄)、浸润性或贮积性疾病(例如淀粉样变性或 Fabry 病)或多系统疾病(例如 Noonan 综合征)所致。
成人 HCM 诊断的典型阈值是最大壁厚度至少 15 mm。对于有 HCM 家族史的患者,特别是携带致病性肌节变异体的患者,13 mm 以上的厚度通常就足够诊断了。然而,这些二元阈值多少有些武断,并未考虑例如性别、体型或合并症等影响因素。
这可能导致过度诊断和漏诊。例如,对于体型偏大且伴有高血压的个体,尤其是在长期高血压控制不好,或者药物治疗效果不佳的情况下,导致持续的压力过载,则可能需要更高的壁厚度诊断阈值。相反,对于女性和身材较小的成年人,则可能需要较低的阈值或将左心室壁厚度与体型关联。此外,心脏形态是可变的。肌节变异最常导致非对称性室间隔肥厚,但最大左心室壁厚度的位置可能出现在其他区域,包括心尖部,或呈同心性(图 1)。
仅关注左心室壁厚度也未能捕捉到 HCM 的完整表型谱和复杂性。改进诊断算法,纳入此类患者特异性特征,以及结合先进影像分析和基因检测,是提高诊断准确性的优先事项。由于这些局限性,左心室厚度评估应结合详细的家族史以及评估 HCM 的其他相关症状,例如心电图异常、二尖瓣和亚瓣膜异常、心肌纤维化和舒张功能异常。
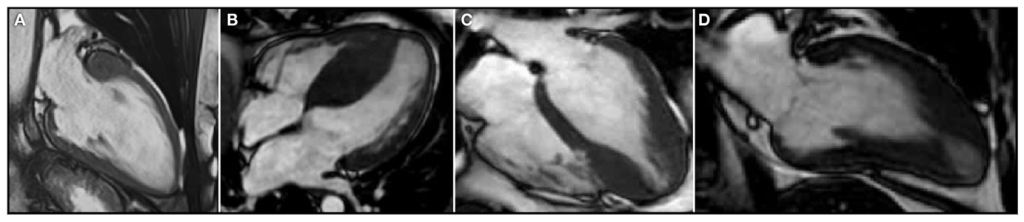
图1. 心脏磁共振成像Cine图像中HCM的表型异质性,两腔视图(A和D)和四腔视图(B和C)。(A) 局部基底隔膜肥厚。(B) 非对称隔膜肥厚。(C) 局部顶端肥厚。(D)中顶端肥厚,伴有中室狭窄和顶端动脉瘤形成初期。
HCM的遗传学基础
成人HCM的患病频率估计为1:500,全球范围内。HCM的单基因病因最初是通过在大型家庭中进行连锁分析而确定的,这些分析显示了多个受影响家庭成员中罕见变异的显性遗传。第一个被确定的HCM基因是MYH7,编码心脏β肌球蛋白重链。如今,编码肌节蛋白的8个基因中的致病变异导致了> 90%的孟德尔遗传病例。在肌节基因中,> 80%的疾病归因于MYBPC3和MYH7中的变异。有少量位于肌节之外的基因,有中等至强的致病性证据,占整体病例的较小比例(表1)。
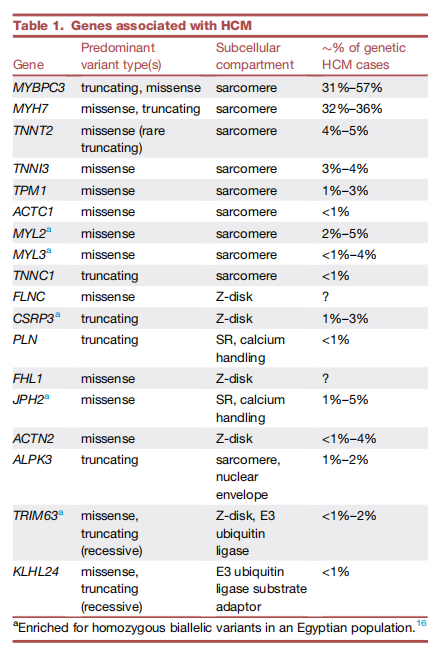
表1 与肥厚型心肌病相关的基因
许多曾经被认为是导致HCM的候选基因已被否定或仍存在疑问,因为这些变异的频率高于疾病患病率,变异在疾病队列中与普通人群相比没有富集,或者它们未能证明家族中遗传变异的连锁。致病性遗传变异的分布因研究人群而异,部分受特定地理区域(如美国、英国)创始人突变的频率影响。与主要来自美国和英国的欧洲/白人血统人群相比,来自新加坡和埃及的患者队列显示,编码肌节蛋白相关蛋白的基因中的变异频率更高,埃及由于近亲结婚,纯合子病例比例更高。
HCM的基因型-表型相关性
不同基因型的HCM患者在疾病进展程度、心脏形态和不良临床结局方面存在显著重叠。携带肌节蛋白变异相关的HCM患者通常比非肌节蛋白变异相关的HCM患者病情发展更恶化,疾病发病年龄更早,房颤、室性心律失常、心力衰竭和死亡的终生风险是其2倍。携带意义未明的肌节变异(VUS)的患者似乎具有中间型表型,可能代表VUS队列中致病性和良性变异的混合,尽管其中一些也可能导致中间临床后果(图2)。
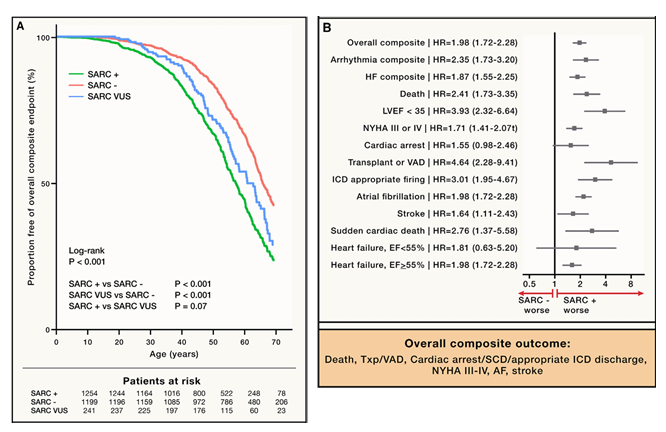
图 2. 肌节突变与临床结局之间的关联
(左)在基因分型的心肌肥厚症患者组进行 Kaplan-Meier 生存分析。与没有肌节突变的 HCM 患者相比,肌节突变携带者 (SARC+) 不良结局发生得更早,发生率也更高,特别是那些携带致病或可能致病变异的患者。到 50 岁时,SARC+ 患者发生综合终点风险为 29.1%,携带 VUS 的患者风险为 24.9%,无肌节突变组患者的风险为 14.2%。
(右) 森林图显示综合终点及其各个组成部分的相对危险度 (HR),比较肌节突变组 (SARC+)、无肌节突变的 HCM 患者 (SARC-) 和携带 VUS 的患者 (SARC VUS) 组。
由于临床和遗传异质性显著,变异特异的基因型-表型相关性在很大程度上并未得到证实,相关的致病变异的识别很少驱动临床决策。
然而,MYH7变异携带者平均发病年龄更早,与MYBPC3变异携带者相比,心力衰竭和房颤的终生风险是其2-3倍。Thin filament基因变异携带者往往表现出较轻的肥厚和较少出现左室出口梗阻,但可能经历向心室收缩功能障碍或限制性生理的心力衰竭进展率更高,尤其是在儿童期发病的患者中。此外,复合杂合子或纯合子与不良结局(特别是不良重塑和左室收缩功能障碍)发生率较高相关。
常见变异和多基因风险的影响
最近的基因组全关联研究(GWAS)病例对照研究,总计4500例病例,发现了与肥厚性心肌病(HCM)相关的多个常见基因位点。多基因风险评分(PRS)与肥厚性心肌病(HCM)诊断几率增加相关,无论是否存在罕见肌节变异。左心室结构/功能与心肌病也存在遗传关联,收缩力增强与HCM风险基因相关,收缩力减弱与扩张型心肌病(DCM)风险基因相关。这些风险基因对HCM和DCM的相反作用,以及与左心室特征(如左室射血分数(LVEF)和应变)的相关性,支持多基因调控收缩功能是心肌病发病机制的核心。
值得注意的是,GWAS研究中发现的大多数基因位点都位于此前与HCM无关的基因中。未来对推定的遗传修饰因子进行临床和实验研究,可能解决与临床风险分层、新疾病途径的发现以及潜在治疗靶点的识别相关的未解决问题。除了遗传修饰因子外,孟德尔随机化研究表明HCM与高血压,特别是收缩压,以及肥胖之间存在因果关系。这突出了在HCM患者中管理合并症的重要性。
重要的是,认识到大多数患者的遗传方式为多基因而非单基因遗传,对家族筛查具有重要意义。对于非家族遗传性HCM患者(即无家族史且遗传检测未发现因果变异),遗传易感性较低,可以考虑对家属进行有限的筛查时间,而不是对有家族史且患病风险更高的亲属进行终身筛查。
疾病机制
肌节收缩力的增强,通常被称为“超收缩性”,以及舒张不足,导致过大的能量消耗,被认为是肥厚型心肌病 (HCM) 的根本生物物理异常。这可能是由于单个肌球蛋白分子的内在力增强、肌球蛋白头部与肌动蛋白结合的易位性增强,或肌球蛋白与肌动蛋白结合的比例增加所致。来自 HCM 的临床前细胞和动物模型,以及具有 MYH7 或 MYBPC3 截短变异的人 HCM 心脏组织的数据显示,在心脏循环的舒张阶段,肌球蛋白分子处于活跃且不完全舒张状态的相对比例增加。这种收缩力增加直接受到新型心脏肌球蛋白 ATPase 变构调节剂的作用,该调节剂会降低横桥的形成率。虽然 MYH7 或 MYBPC3 中的致病变异导致肌球蛋白活化驱动了 HCM 的病理生理学,但该机制与其他 HCM 病因的相关性尚不明确。
在直接影响收缩性的生物物理效应下游,在 HCM 患者心脏组织(包括心脏切除标本和心脏移植或死亡时取出的心脏)中,已经鉴定了其他疾病通路。最近的无偏“组学”方法显示,许多通路(例如能量代谢、钙稳态和蛋白质折叠)下调,以及参与细胞外基质重塑、炎症反应和细胞骨架重组的通路上调。通过研究 HCM 心脏组织,已经鉴定了依赖基因型和不依赖基因型的信号通路。这些通路也参与了扩张型心肌病 (DCM) 的发病机制,且作用方向可能协同或拮抗。由于通常在疾病晚期才能获得人体心脏组织,这些数据表明心室重塑晚期存在共同点。
临床病程和预后
HCM 的诊断年龄从儿童期到超过 60 岁不等。肌节蛋白相关的 HCM 疾病早期发作更为常见,并且是不良事件的强有力预测指标。然而,临床表现差异很大,从轻微症状、相对正常的预期寿命到晚期心衰或恶性心律失常都有可能。随着时间的推移,主要表现为心房颤动和症状性心力衰竭,通常出现在中年,并且通常在临床显性表型首次出现数年至数十年后。大约 8% 的患者出现左室收缩功能下降(左室射血分数 < 50%),通常伴有广泛的心肌纤维化。这种高级重塑非常不利,会增加晚期心力衰竭、机械辅助或心脏移植以及死亡的风险。患有 HCM 的患者在任何年龄都可能发生危及生命的室性心律失常,但年轻人风险最大。儿童期猝死性心律失常的可能性约为每年2%,成人约为每年1%,直到 70 岁以后,心律失常事件很少见,尤其是非肌节蛋白型 HCM。
基因检测的日益普及有助于识别基因阳性、左室肥厚阴性(基因阳性,左室肥厚阴性)的个体,他们存在临床前 HCM。这些个体遗传了导致其家族中 HCM 的致病性肌节蛋白变异,但左室壁厚度正常,不符合疾病诊断标准。临床前 HCM 几乎没有不良事件。由于肌节蛋白变异的遗传易感性不完全是,只有大约 60% 最终会转化为明显的 HCM,通常发生在生命的前四十年。然而,在左室肥厚发生之前,可以检测到肌节蛋白变异的早期征象,包括左室舒张功能受损、纤维化倾向增强、心肌能量代谢改变、僧帽瓣和左室几何结构异常、以及ECG异常。这些特征的存在可能识别出患有更高风险的个体,可能很快发展为临床显性疾病,尽管目前无法预测表型转换的可能性或时间。
识别出患有亚临床或早期阶段HCM的个体,为启动改变疾病进程的疗法提供了机会,这些疗法可以在建立广泛的、可能不可逆的室性重塑之前减缓表型进展。先前的动物和人体研究表明,药物治疗可以减轻不良重塑。“早期肌节蛋白型 HCM 中Valsartan减缓疾病进展”(VANISH)试验表明,Valsartan对早期 HCM 的年轻健康携带者有益。与安慰剂相比,Valsartan治疗与心脏重塑综合指标的稳定或改善有关,该指标整合了左室结构功能和心脏生物标志物。值得注意的是,在左室肥厚不严重的参与者中,益处最大,支持了疾病修正可能在存在高级重塑之前更成功的假设。Valsartan对临床结果(如心律失常或症状负担)的影响并未进行测试,因为需要很长的时间和大量参与者。一般而言,在早期或亚临床疾病中进行试验面临着 HCM 的自然病程复杂且漫长 的挑战。为了开发有效的疾病修正疗法,迫切需要识别能够反映表型进展并可用于监测新型治疗反应的可靠生物标志物。
临床管理个性化进展
目前,治疗的目标是控制症状。β受体阻滞剂或非二氢吡啶类钙通道阻滞剂一直是首选的药物治疗,但它们尚未经过系统性研究,许多患者并未体验到足够的症状缓解,或伴有显著的副作用。如果认为阻塞性生理因素是导致症状的原因,那么就会考虑进行侵入性的隔膜减容治疗,如手术隔膜心肌切除术或基于导管的酒精隔膜消融术。
心肌肌球蛋白抑制剂(CMIs)是针对肥厚型心肌病(HCM)开发的第一种疾病特异性治疗方法。这些心脏肌球蛋白的变构调节剂是基于观察超动态收缩作为肌节变异的基本表现而开发的。具体药物包括Mavacamten(马瓦卡坦/玛伐凯泰,2022年在美国获得治疗症状性梗阻性HCM的批准,2023年由欧洲药品管理局批准)和Aficamten(2023年完成相同指征的3期临床试验)。
Mavacamten与肌球蛋白结合,降低无机磷酸盐的释放速率,稳定肌球蛋白的完全松弛、低能耗构象。Aficamten具有类似的药理机制,尽管与Mavacamten相比,它可能稳定不同的肌球蛋白非活性状态。在梗阻性HCM患者中使用CMIs的随机临床试验一致显示,左心室流出道(LVOT)梯度显著降低,运动能力和生活质量改善,循环中N端前B型利钠肽(NT-proBNP)和高敏肌钙蛋白水平降低,以及对舒张功能的益处。
纵向分析表明,左心室质量(LV)和左心房(LA)的大小都有所减少。这两种药物都显示出良好的安全性和耐受性。作为肌球蛋白抑制剂,它们会降低左室射血分数(LVEF),但影响通常较小。然而,在临床研究和使用Mavacamten治疗阻塞性肥厚型心肌病(HCM)的患者中,观察到高达7%-10%的患者LVEF低于50%,这导致剂量减少或停药。因此,在治疗开始和长期使用过程中,需要仔细监测LVEF,这遵循FDA强制实行的风险降低策略(REMS)以及其他类似计划。
治疗无阻塞性生理学的症状性患者仍然具有挑战性,目前尚无有效的治疗方案。鉴于CMI改善舒张功能和心肌能量代谢的潜力,非阻塞性肥厚型心肌病的假设性驱动生理路径,Mavacamten和Aficamten目前正在进行症状性、非阻塞性HCM患者的随机3期临床试验。除了症状缓解之外,人们对CMI可能能够改变疾病轨迹持乐观态度。在HCM小鼠模型中,在LVH发生之前给予mavacamten可以预防肥厚和纤维化的发生。在症状性HCM患者中执行CMI疾病修饰的临床试验将首先需要长期安全性的稳健证据。此外,还需要创新策略来识别疾病进展的可靠生物标志物,以及监测早期无症状HCM的治疗益处。
除了CMI外,还有其他小分子药物正在研发,以靶向治疗HCM。EDG-7500是一种首创的口服心脏肌节选择性调节剂,旨在减慢早期收缩速度,改善心肌舒张,适用于肥厚性心肌病患者。临床前数据显示,该药物在梗阻性肥厚性心肌病动物模型中有效,且对收缩功能影响不大。目前正在进行EDG-7500的开放标签剂量寻找试验(NCT06347159)。初步短期结果显示,该药物显著降低了流出道梯度和心肌生物标志物水平,而左室射血分数(LVEF)变化不大。
另一种线粒体代谢调节剂ninerafaxstat,通过部分抑制脂肪酸氧化,并直接竞争性抑制3-酮酰辅酶A(CoA)硫解酶(3-KAT,线粒体长链脂肪酸β-氧化途径中的关键酶),将心脏代谢转向糖酵解,发挥作用。Ninerafaxstat可以提高心脏效率,降低产生每摩尔ATP所需的氧气量。在涉及非梗阻性肥厚性心肌病患者的2期研究(NCT04826185)中,服用ninerafaxstat的患者通气效率有所改善,但峰值VO2和症状评分两组之间无统计学差异。正在计划进行3期研究,以测试该药物在更大样本量非梗阻性肥厚型心肌病患者中的疗效。
另一类可能对HCM患者有益的药物是钠-葡萄糖协同转运蛋白抑制剂(SGLT抑制剂)。这类药物已显著降低了心力衰竭患者(包括射血分数降低和保留的患者)的死亡率和住院率。虽然其作用机制尚不清楚,但临床前证据表明,它可以直接加速舒张,提高利用率。正在进行针对梗阻性和非梗阻性肥厚性心肌病患者的临床试验(sotagliflozin,参与SONATA-HCM [NCT0648189]和SOTA-CROSS [NCT06433050];empagliflozin,参与EMPA-REPAIR [NCT05182658])。
扩张型心肌病(DCM)和心律失常型心肌病(ACM)
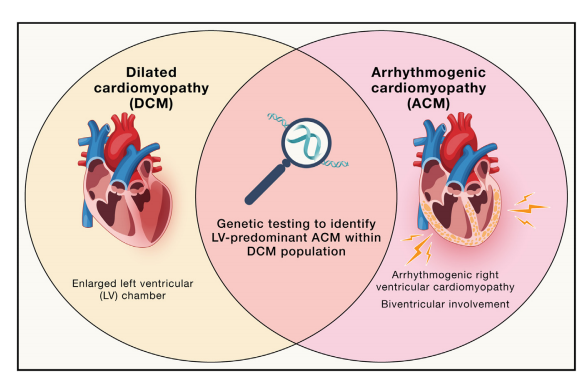
图 3. DCM 和 ACM 的表型重合,基因检测有助于区分 DCM 患者中的左室为主的ACM。
目前的共识声明表明,初始临床表现为心律失常,以及特定的影像学特征,提示ACM 的诊断。ACM包含左室为主和右室为主的各种心肌病。左室为主的 ACM 可能被误诊为原发性 DCM。
DCM与ACM定义
扩张型心肌病(DCM)和心律失常型心肌病(ACM)是复杂的疾病,通过扩大心腔、降低收缩力来改变心脏结构和功能。历史上,DCM被认为是左心室疾病,心律失常程度变化较大;而ACM主要描述右心室疾病,伴有纤维脂肪沉积和致命性室性心律失常(最初被称为右室心肌病[ARVC],定义于2010年工作组标准)。然而,在扩张型心肌病患者中,早期出现轻度心室扩大和收缩功能下降的致命性室性心律失常,需要修改这些定义。现在,DCM是指以左心室扩大和收缩功能下降为主的疾病;而ACM则泛指影响右心室、左心室或双心室,且致命性室性心律失常程度与其收缩功能下降不成比例的心肌病(图3)。
基于遗传病因而不是临床表现来分类,可以建立更精确的分类,并深入了解疾病自然发展,这有助于风险评估和治疗方案制定。因此,将命名方法从形态学转向遗传学将更有临床价值。
遗传基础:DCM和ACM
DCM和ACM的基因检测阳性率低于HCM。DCM病例中,致病或疑似致病变异检出率为25%至35%。尽管尚未对ACM不同类型(右室和左室)的基因检测阳性率进行量化,但在符合工作组标准的明确ARVC病例中,检出率高达58%。DCM中,提高诊断检出率的临床因素包括合并骨骼肌病变、家族史和无合并症(例如,束支传导阻滞和高血压)。表2总结了导致DCM和ACM的基因、其可能的细胞机制以及基因特异性临床特征。
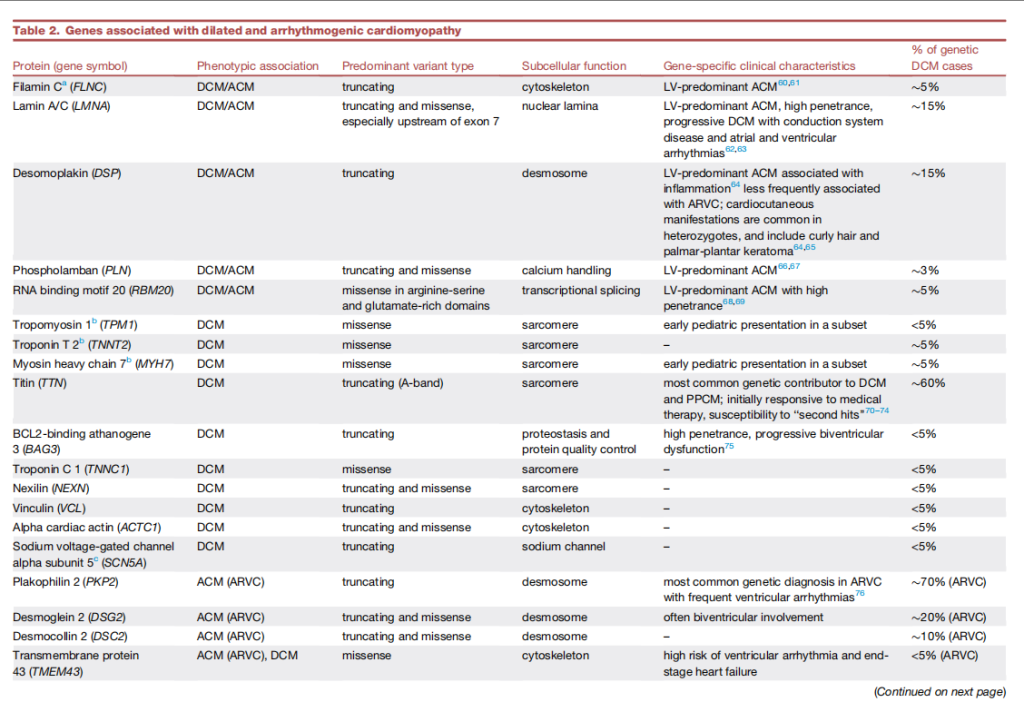

表2. 扩张型和致心律失常型心肌病相关基因
细胞骨架
中间纤维、肌原纤维和核膜对于维持心肌细胞的机械转导、肌节排列和成熟心脏转录程序至关重要。这些分子结构的破坏与具有特定特征的心肌病有关。例如,LMNA基因的变异,常伴有危及生命的室性心律失常、房性心律失常、传导系统疾病、左室扩张以及核结构紊乱。最近的研究表明,层蛋白-染色质相互作用的破坏会导致非心肌基因表达,并损害正常心肌功能。LMNA与另一种核因子跨膜蛋白43(TMEM43)结合,其中错义变异与ARVC有关。
纤连蛋白C (FLNC) 的截短变异被认为会破坏它在细胞内肌动蛋白与细胞外基质之间连接桥梁的作用,从而导致ACM;这导致蛋白质稳态受损,并加速自噬。相反,FLNC杆状结构域的错义变异导致蛋白质错误折叠,从而导致肌纤维病伴心肌肥厚。DES的致病变异也会导致错误折叠蛋白聚集,形成可溶性淀粉样前寡聚体,从而破坏肌肉功能,导致肌纤维病,影响骨骼肌和心肌。
DMD和EMD基因的变异会导致 X 连锁肌营养不良和早期进行性扩张性心肌病。EMD 对核膜结构至关重要,DMD 是一种关键的膜稳定剂,它维持肌细胞的细胞内-细胞外连接,并参与对肌原纤维结构和钙负荷至关重要的信号传导。DMD 的截短变异会导致严重的疾病,即杜氏肌营养不良症 (DMD),这通过单倍体不足机制发生。贝氏肌营养不良症 (BMD) 通常由 DMD 的错义突变或非同义突变引起,这些突变导致功能部分缺失,但未完全导致单倍体不足现象,并具有较轻的临床表型,尽管 DMD 和 BMD 相关的疾病严重程度谱存在重叠。一项最近对Emery-Dreifuss型肌营养不良 (EDMD) 患者的登记发现,EMD (EDMD1) 的变异与 LMNA (EDMD2 和 3) 变异引起的变异一样具有侵略性,并建议尽早考虑心脏衰竭治疗,并进行植入式心脏除颤器的预防性植入。
桥粒是细胞间粘附和通讯的关键结构。一些桥粒基因(例如, PKP2和 DSG2)的变异会导致右心室心肌病,伴有心肌纤维脂肪浸润。桥粒蛋白 (DSP) 的截短变异也导致心肌病 (主要累及左室),并与 15% 的病例中的炎症反应有关。心肌病的确切分子基础尚不清楚,但可能涉及炎症反应以及由于细胞间通讯中断和损伤易感性而改变的纤维化信号。纤维脂肪浸润被认为源于异常的β-连环蛋白信号传导,以及其他促脂肪生成刺激,以及持续的心内炎症,可能由抗桥粒抗体介导。心肌细胞死亡和纤维脂肪替代的其他建议机制包括蛋白质质量控制受损,膜整合素的相互作用异常和新型转录因子激活。
横纹肌
横纹肌蛋白基因中的致病性变异也可以导致扩张型心肌病(DCM)。这些DCM变异与导致肥厚型心肌病(HCM)的变异不同,且比后者更为罕见。MYH7中的DCM错义变异会降低横纹肌的收缩力生成,导致扩张性低收缩表型,与增加收缩力的HCM变异不同。DCM还可能由肌钙蛋白T(TNNT2)和肌钙蛋白C(TNNC1)的截短或错义变异引起,以及不引起HCM的横纹肌相关蛋白,包括 nexilin(NEXN)和 肌联蛋白titin(TTN)(表2)。
TTN 是一种巨型支架蛋白,作为肌节的分子弹簧。TTN 基因的截短变异(TTNtvs)是扩张型心肌病(DCM)最常见的遗传原因,占所有 DCM 病例的 10% 至 20%。值得注意的是,TTNtvs在普通人群中相对普遍。尽管扩张型心肌病的明显特征可能不存在,但普通人群中发现的 TTNtvs 会导致与非携带者相比的室壁形态的微妙变化。高频转录的TTN外显子中发生TTNtvs(高剪接率)更有可能具有致病性,并且与心力衰竭患者的预后较差有关。TTN 的长度和磷酸化变化调节左室舒张功能,包括扩张性心肌病和射血分数保留的心力衰竭 (HFpEF)。该基因 A-band的 TTNtvs(与人类扩张型心肌病高度相关)被翻译,似乎会破坏正常的肌节功能。截短的TTN蛋白可能整合到肌节中,或者可能损害肌原纤维形成,这意味着肌节功能障碍可能源于单倍体不足和显性负面效应。相关的肌节紊乱可能会诱导收缩功能和肌原纤维组织的缺陷,从而降低心肌细胞适应增加的机械负荷或交感神经刺激的能力。
钙离子稳态和离子通道
参与膜电位调控和钙离子循环的基因变异与DCM和ACM相关。钠通道Nav1.5(SCN5A)电压门控结构域的错义突变与ACM相关,据认为其降低电压门控钠离子电流,从而降低收缩力,并导致钙诱导的钙释放。收缩结束后,钙离子从细胞质重新摄取到肌质网,通过SERCA进行,其结合PLN进行调节。PLN对肌质网钙储存的失调是致病性的,无论是功能缺失或功能增强变异都会导致ACM。两种变异导致DCM的机制尚不完全清楚,但细胞内钙库和细胞质钙水平的稳态对于维持心律和舒张功能至关重要。
RNA表达、剪接和蛋白质质量控制
心肌细胞是高度分化的终末细胞,不具备分裂能力,对蛋白稳态和氧化应激极其敏感,依赖于严格的基因表达和剪接控制,以快速更新肌节和T管蛋白。影响这些过程的多个基因的变异是DCM和ACM的罕见原因。例如,RBM20调控参与心肌兴奋收缩耦联的基因剪接,特别是肌节蛋白和钙调节蛋白(例如,TTN、CAMK2D、LDB3和RYR2)。BAG3的致病变异被认为通过破坏肌细胞中的自噬和蛋白质稳态导致DCM和肌球蛋白病。
基因型-表型相关性与DCM/ACM的临床意义
传统DCM诊断需要左心室收缩功能障碍(射血分数低于50%)和/或左心室扩张超过正常范围的95百分位数。历史上,我们对DCM诊断和自然病史的理解并未充分考虑潜在的病理机制。然而,基因诊断可以识别高风险人群,并且这些患者可能在诊断标准明确之前或仅伴有轻微的左心室结构改变时就出现疾病的早期表现。例如,LMNA、DSP、PLN、RBM20和FLNC等基因的致病突变经常导致患者在临床上表现为轻度DCM的情况下出现早期危及生命的室性心律失常。相反,TTN突变相关的DCM患者与其他DCM患者相比,早期危及生命的室性心律失常风险较低,但其室性心律失常风险高于普通DCM患者。
特定的基因型与高度易发生心律失常的左心室为主的收缩功能障碍表现相关。DSP基因的致病突变可导致反复发作的心肌炎。FLNC基因的致病突变同样可以引发在DCM症状明显之前的心脏炎症和心律失常。PLN心肌病的自然病史也类似。国际合作促进了针对多种心肌病基因,包括PLN、DSP和LMNA,开发特异性室性心律失常风险预测工具。致病性LMNA突变具有高度外显率,并导致独特的室性心律失常谱,伴有进行性房室传导系统损害,通常需要起搏器和植入式心脏复律除颤器(ICD),通常在DCM症状明显之前。更高的心律失常风险与截短突变(相对于错义突变)、男性性别以及间歇性室性心动过速或左心室射血率低于45%的个体相关。(图4)
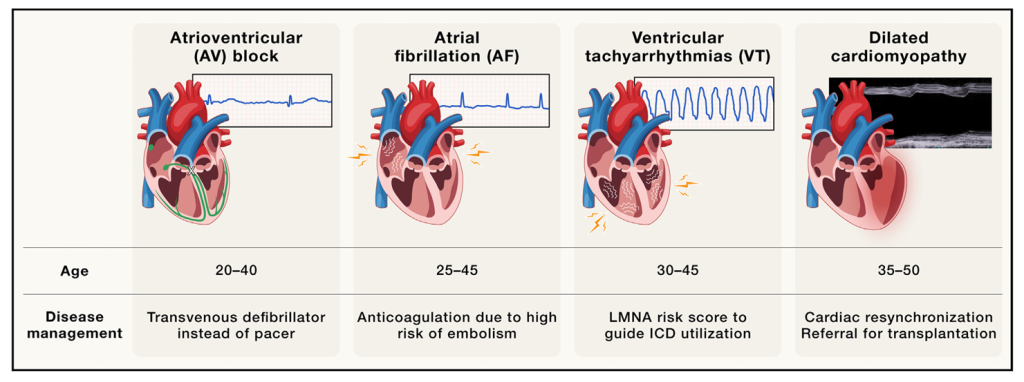
图4. LMNA心肌病的疾病发展过程指导临床实践
LMNA心脏病中,房室传导阻滞、心房颤动、室性早搏和扩张性心肌病的年龄相关性发病率。最后一行描述了针对LMNA心肌病的具体治疗方案,突出了基因诊断的重要性。
TTNtvs的临床表现包括原发性心肌病,但它们也可能增加与多基因风险或基因-环境相互作用相关的“第二次打击”而发展为扩张型心肌病(DCM)的易感性。在学术医疗中心的基因分型队列中,与非洲血统相比,TTNtvs引起的DCM在具有欧洲血统的患者中侵袭性较低。相反,TTNtvs与妊娠、心脏毒性癌症化疗和酒精摄入相关,增加了DCM的风险。这些观察结果表明,基因型结合行为改变和医疗干预有可能降低发展为DCM的风险。
治疗标准
DCM和ACM的治疗旨在减轻症状、心律失常和进展为心力衰竭。识别猝死风险也是临床护理的重要方面。由于恶性心律失常风险增加,指南和共识声明认为,存在致病性LMNA、FLNC、TMEM43、PLN、RBM20和DSP变异是考虑进行一级预防性植入心脏除颤器(ICD)以预防猝死的指征,即使在没有传统危险因素或严重左心室功能不全(LVEF < 35%;这是DCM植入的标准指征)的情况下也是如此。这些发现也对家庭有影响,因为它们可能促使对有风险和受影响的亲属进行更积极的管理,例如更频繁的心律监测和对诸如非持续性室性心动过速等临床风险的警惕,这些风险在传统的DCM患者或具有正常左室收缩功能的LMNA变异个体中可能被轻视。LMNA心肌病中潜在的致命性心律失常可能在明确的结构表型出现之前发生。因此,对家庭成员进行级联基因检测对于决策至关重要,因为单独的致病性LMNA变异就足以建议加强心律失常监测和生活方式干预。
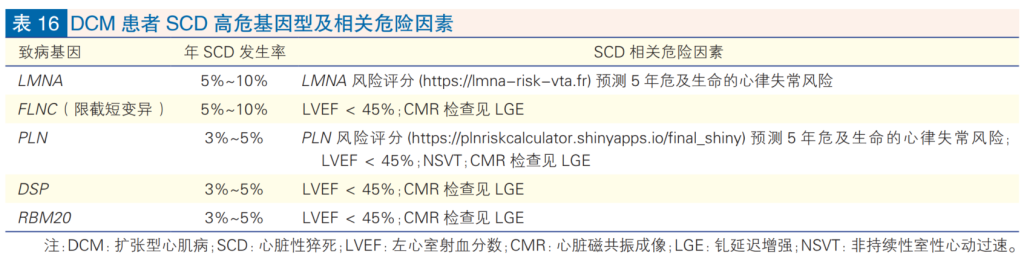
《中国心肌病综合管理指南 2025》中DCM患者SCD高危基因型
过去十年进行的大型随机临床试验已为左心室射血分数降低的心力衰竭患者建立了循证指南下的药物治疗(GDMT),包括β受体阻滞剂、醛固酮受体和神经激肽酶抑制剂、盐皮质激素受体拮抗剂和SGLT2抑制剂。GDMT已降低了大规模DCM患者群体死亡率和发病率,并减缓了收缩功能的下降。
但是,这些疗法的成功,特别是针对遗传性心肌病,是可变的。例如,TTNtv DCM通常对室壁去负荷和GDMT相对敏感,尽管在初始改善后,射血分数可能随着时间推移而缓慢下降。此外,TTNtvs在早期发作的心房颤动患者群体中富集,包括那些未确诊为心肌病的个体。LMNA心肌病患者可能对GDMT的反应较弱,但是,最近的证据表明,这些患者对心脏同步化疗法(CRT)有反应,特别是那些LVEF较低且室性起搏百分比较高者。目前尚无循证医学疗法支持ARVC中的右心室收缩功能障碍。尽管已经研究了基因型特异性DCM的小分子精准疗法,但均未显示出足够的益处以达到临床应用水平。
核酸药物:下一代治疗策略
核酸药物将RNA或DNA递送至针对导致疾病的致病基因型产生的事件。这些疗法通过提供外源RNA或DNA序列进行转录和翻译,或改变基因组的序列、转录或翻译,从而介导瞬时或长期的细胞效应。虽然曾经是遥远的目标,但核酸药物开发和递送方面的快速进步正在推动遗传性心肌病的首例人类临床试验。
尽管在多种HCM、DCM和ACM基因中存在数百种致病或潜在致病变异,但核酸药物利用了致病变异通常产生两种机制后果的证据:显性负效应或单倍体不足症。这些疾病机制需要不同的治疗策略,如图5所示。
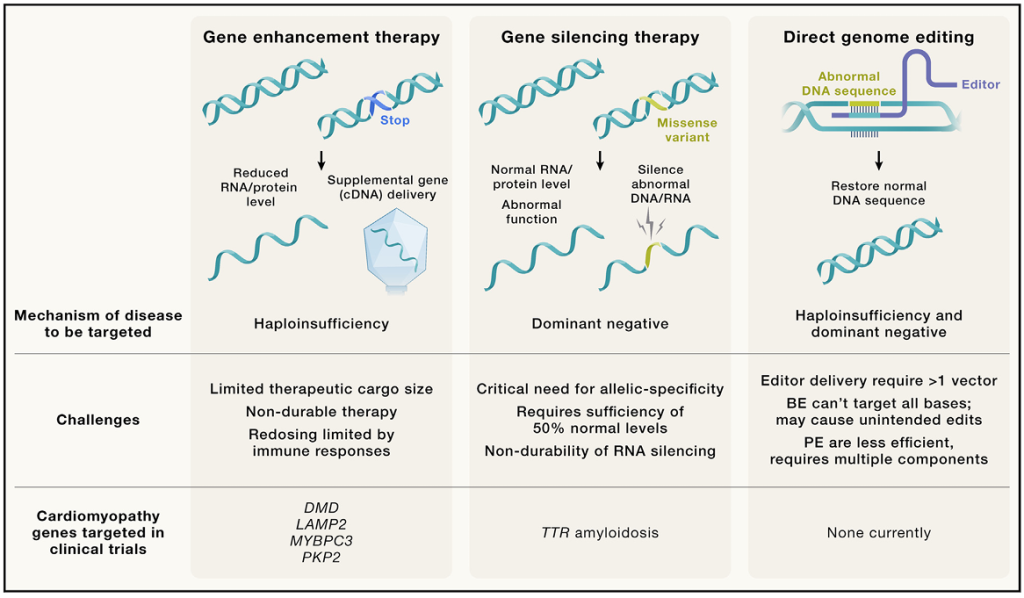
图 5. 基因治疗策略包括基因替代、基因沉默和基因组编辑。例如,在功能缺失或单倍不足模型中,基因替代提供基因的功能性拷贝。基因沉默降低那些基因变异编码了毒性蛋白的基因表达量。基因组编辑旨在直接编辑基因组以纠正致病变异或敲除编码毒性蛋白的基因。
基于编辑或沉默策略应对显性负效应
显性负效应(Dominant negative)变异产生错义突变或同义突变(插入或缺失),改变了产生的蛋白质的功能,并干扰了正常的细胞过程。例如,致病性MYH7错义突变被认为通过破坏肌节蛋白的正常相互作用,导致舒张不充分,并促进过度收缩,从而引起肥厚性心肌病。目前正在研究的治疗显性负效应心肌病的方法包括将小非编码干扰RNA(RNAi)递送至心脏,以暂时性且选择性地沉默突变转录本,以及递送碱基编辑器或核酸酶,并带有等位基因特异性向导以永久性地纠正或失活突变DNA序列。
要使基因沉默方法可行,必须有明确的证据表明,产生的单个功能性等位基因足以满足编码蛋白的所有生理和应激诱导需求,并且产生的单倍体不足本身不会导致疾病。涉及直接DNA编辑的策略必须限制意外的旁观者突变和脱靶突变,这些突变可能会产生新的有害变异。使用组织特异性启动子,将表达限制在靶向细胞中,可以部分减轻这些负面影响,但这些措施可能并不完美。此外,尽管心肌细胞缺乏复制能力降低了致癌风险,但在其他组织中低水平摄取和表达的可能性提出了是否需要对治疗患者进行长期癌症监测的问题。
数千种不同的错义突变会导致心肌病,因此逐个纠正每一种变异在实践中是不可行的。另一种方法是通过靶向区分突变等位基因和健康等位基因的常见单核苷酸多态性(SNP)来选择性地沉默突变转录本。对新生小鼠进行处理,使用靶向仅存在于携带HCM MYH7变异Arg403Gln的突变转录本上的SNP的RNAi,其疗效与直接沉默致病变异一样好。6个月后,肥大和心肌纤维化出现,之后RNAi水平下降,随后出现HCM。这种方法说明了在人群中常见且选择性地针对突变等位基因的SNP如何克服每个显性负性变异设计独特构建体的困难。此外,尽管治疗效果是暂时的,但这些分析表明,降低突变转录本25%足以预防疾病——这一观察结果暗示了低剂量重复治疗的潜在治疗效果。siRNA对突变等位基因的选择性至关重要,以避免引起单倍体不足,因为许多心肌病基因对剂量敏感。
碱基编辑器已在小鼠中永久性地纠正了HCM变异。这些通常采用腺相关病毒9(AAV9)载体和心肌细胞特异性启动子以靶向递送并限制心肌细胞中碱基编辑器的表达。碱基编辑器直接修饰核苷酸,而不会将潜在的有害双链断裂引入DNA中。胞嘧啶编辑器将胞嘧啶转化为胸腺嘧啶(或鸟嘌呤转化为腺嘌呤)核苷酸。腺嘌呤编辑器将腺嘌呤转化为鸟嘌呤(或胸腺嘧啶转化为胞嘧啶)核苷酸。AAV9的载荷量有限,需要使用双载体,每个载体都编码碱基编辑器的一半,并带有RNA向导,融合到反式剪接裂解内切酶的一半,以便在转染细胞中重建编辑器。
虽然需要进一步研究以检查低水平脱靶突变的潜在长期影响,但这些研究暗示了在疾病显现之前纠正错义突变以预防遗传性心肌病的潜力。碱基编辑器以类似的方式用于治疗ACM中的显性负效应变异。但是,由于腺相关病毒(AAV)的载荷量有限、心肌血管对脂质纳米粒子的相对不渗透性以及旁观者编辑和脱靶效应的可能性,直接对人心脏进行基因编辑仍然具有挑战性。目前尚未启动心肌病基因沉默或碱基编辑的临床试验。
单倍体不足变异的基因置换策略
单倍体不足变异通过引入提前终止密码子、破坏剪接或编码导致蛋白质不稳定,并降低细胞所需蛋白质量的错义突变来引起疾病。对于单倍体不足变异,当前策略旨在递送功能性转基因或纠正或修饰突变基因以产生全功能或部分功能蛋白质(图 5)。第一种基因置换策略是递送非永久性功能性转基因,不改变患者的天然 DNA 序列,在临床开发中处于领先地位。这种方法适用于 MYBPC3 基因的失功能突变,这是最常见的遗传性肥厚性心肌病 (HCM) 原因,以及 DCM 和 ACM,它们也多由导致单倍体不足的突变引起。
基因置换可以通过包装外源性野生型 cDNA,并通过 AAV9 载体递送来实现。这种策略已在丹农病中获得早期经验,丹农病是一种致命的 X 连锁疾病,由 LAMP2 基因的失功能突变引起,该基因编码参与细胞自噬的关键的小型溶酶体相关膜蛋白。丹农病会导致肝功能障碍和进行性、高度 ACM,伴有极度左心室肥厚。早期人类临床试验(使用全身静脉递送)因药物不良反应而面临挑战,需要降低剂量并使用免疫抑制剂。最近的一期临床试验报告称,接受 LAMP2b 转基因的六名男性患者的疾病有所改善或稳定。在这项以及其他使用 AAV 载体的试验中,可能会出现一些严重的副作用(例如,血栓性微血管病、肾衰竭、肝损伤和心肌炎),这可能是由于载体高剂量和对载体或递送蛋白质的免疫反应所致。
通过胸腔内递送携带 Tnnt2 启动子和 Mybpc3 cDNA 的 AAV9 处理具有 Mybpc3 同型缺失的新生小鼠,蛋白质水平升高,并防止心肌重塑和功能障碍长达 30 周。一项平行研究向同型 Mybpc3 缺失小鼠递送单剂量的完整人 MYBPC3 cDNA 构建体,逆转了心脏表现并改善了 1 年存活率。转基因在健康非人灵长类动物体内耐受性良好,目前正在进行一项针对 MYBPC3 单倍体不足引起的 HCM 的临床试验,该试验正在递增转基因剂量(临床试验.gov 标识符 NCT05836259)。同样地,在由 PKP2 中的截短突变引起的 ARVC 中,三项研究报告称,在 AAV 载体中用 cDNA 构建体替换 Pkp2 挽救了小鼠心脏模型中的斑点蛋白 2 蛋白质表达,并防止了右心室功能障碍和心律失常。目前,正在进行三项临床试验以将这种潜在疗法扩展到人类(NCT05885412、NCT06109181 和 NCT06228924)。
心肌病精准核酸疗法的实现面临挑战
将核酸疗法送达心肌存在挑战。通过发现心脏靶向病毒(例如,AAV9/41),改造AAV衣壳提高心脏特异性,以及使用心肌细胞特异性启动子(例如,TNNT2启动子)来改善递送。然而,AAV介导疗法的最大限制是载荷量小(小于等于5 kb)。这限制了其在小RNA疗法和适当大小的DNA分子中的应用。较大的载荷,例如碱基编辑工具,需要更复杂的策略,例如使用包含裂解内含子的双AAV,促进拼接,重建分子结构。
病毒递送的另一个主要限制是免疫原性。很多人因为之前接触过病毒,体内有抗病毒衣壳抗体,这限制病毒载体进入细胞,引发炎症反应。此外,一次治疗后就会产生中和抗体,阻碍后续治疗。可以通过联合使用免疫抑制剂来缓解这个问题,但这些药物本身也存在风险。
为了让疗法作用于大部分心肌细胞,系统给药可能需要高剂量载体,并可能存在剂量依赖性毒副作用。治疗还可能引发对病毒衣壳或基因治疗系统中细菌成分的严重免疫反应,甚至危及生命。预防性免疫调节可能减轻这些反应的严重性。为了解决AAV的免疫反应和载荷量限制,人们正在开发脂质纳米颗粒、病毒样颗粒(VLPs)和包膜VLPs。脂质纳米颗粒(LNPs)已被用于将基因编辑工具递送到肝脏,以抑制家族性高胆固醇血症中的PCSK9活性。然而,LNPs不易被心肌细胞吸收,可能需要心脏内注射和分子工程来改造增强吸收的表面分子。
除了巨大的技术挑战外,实施心肌病基因疗法还有巨大的临床挑战。不同患者的致病变异临床表现差异很大,即使是同一种疾病基因也是如此。因此,选择合适的患者至关重要。如果等到疾病晚期才治疗,效果可能无法完全恢复功能。对携带致病变异但尚未发病的个体进行干预,可能更有效,但如果发病风险和不良后果不明确,则不合适。目前,生物标志物和影像学检查还不能精确地判断疾病进展和高风险患者。治疗的潜在益处必须权衡急性毒副作用和潜在的长期后果。解决这些根本问题对于识别最有可能受益于明确核酸疗法的患者以及评估治疗有效性至关重要。
核酸疗法应用将对医疗体系和政策带来挑战。基因疗法的初期成本很高,目前每剂可能需要数百万美元。然而,成本效益(以每调整后生命年成本计算)差异很大,对于某些治疗方法,成本与美国合理的成本效益阈值相符。由于治疗人数少,缺乏重复研究,以及疗效持续时间和程度不明确,成本效益研究仍有局限性。公平获得这些疗法将是一个巨大的挑战。
心肌病是导致心力衰竭的主要原因,心力衰竭是一种致命疾病,全球有6430万人受其影响。预计到2030年,每位美国成年人的医疗成本将达到244美元。通过生物工程师、分子科学家、临床医生和医疗保健系统的集体投入,开发安全有效的基因疗法来治疗心肌病将造福患者、家庭和社会。
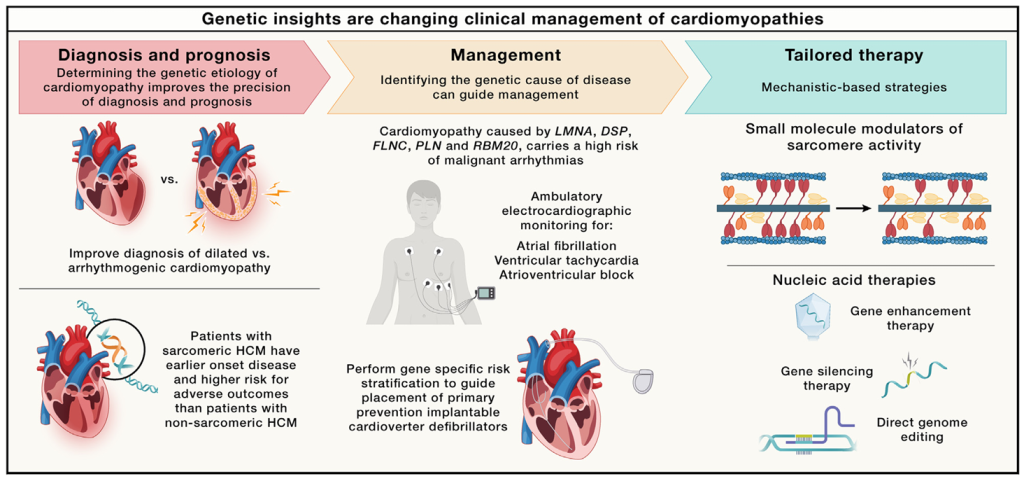
图6. 通过将遗传信息整合到临床医学中,有望实现预后改善、治疗管理优化及创新疗法的开发
结论
研究人类心脏病的遗传基础为理解、预测和预防疾病提供了非凡的机会。遗传性心肌病为实现这一机会提供了范例。遗传学见解可以通过将诊断从基于心脏结构/功能异常转变为基于分子机制,并通过开发靶向潜在病理生物学的治疗方法来改变医学。
越来越多的心肌病患者可以获得遗传诊断,这使得临床模式能够从形态学基础转变为机制基础。遗传诊断的粒度和特异性将病因与表型和临床表现联系起来(图6)。例如,肥厚性心肌病与不良事件的发生率更高有关,包括心力衰竭、室性和房性心律失常以及死亡。由LMNA、DES、FLNC、DSP、PLN和RBM20基因变异引起的心肌病与恶性心律失常的高风险相关,这需要更积极的监测和不同的考虑植入心脏起搏器(ICD)进行一级预防的阈值。然而,由于未能采用全面的遗传诊断以及对不同人群多基因风险的了解有限,仍然有许多患者难以进行基因诊断。需要在生物库和疾病人群中进行扩展测序,以加深理解并为患者提供临床转化信息。
遗传性心血管疾病的个体化治疗正在成为现实。在肥厚性心肌病中,心脏肌球蛋白调节剂是首批开发并被证明能够改善症状负荷和功能能力的针对该病的药物。正在测试使用其他靶向肌节的小分子、SGLT2抑制剂和代谢调节剂的新方法。肌球蛋白激活剂已被提议用于扩张型心肌病,免疫调节剂已被提议用于炎症性心肌病的疾病调节。
尽管距离临床应用尚远,但目前一些心肌病的基因替代疗法试验,正在招募患者。鉴于这些方法的复杂性和风险更大,需要更安全的方法和优化患者选择,才能真正实现临床应用。此外,我们需要确定给这些新疗法安全施用(或投递)的最佳时机。更早治疗是否会带来更显著的效果?治疗效果是否持久?症状和猝死风险达到何种程度,可能超过新疗法的风险?我们有什么信息可以指导与患者的共同决定?尽管遗传性心脏病仍需更多进展,但基因型特异性风险分层和管理方面的成就,让大家期待已久的未来,现在变成了现实。
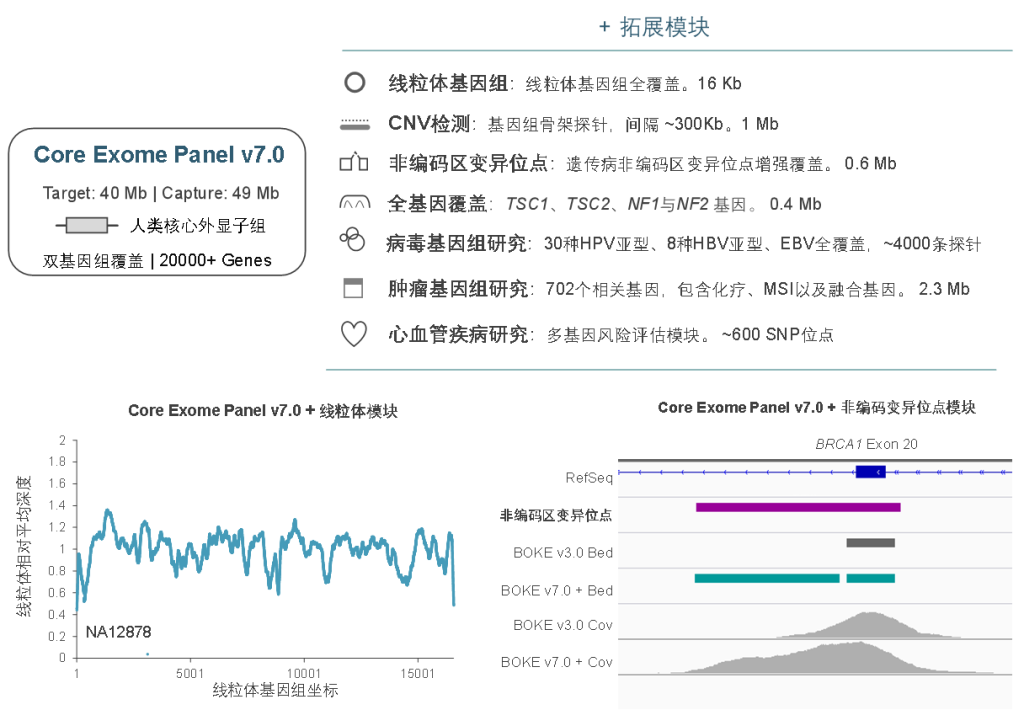
TargetCap® Core Exome Panel v3.0
TargetCap@ Core Exome Panel v3.0基于伯科高品质DNA探针合成技术开发,全流程国产制造,由~40万条探针组成,以GRCh38/hg38人类参考基因组设计,参考Refseq、CCDS、ClinVar等数据库,覆盖19,524个基因,目标区域为33.9Mb。
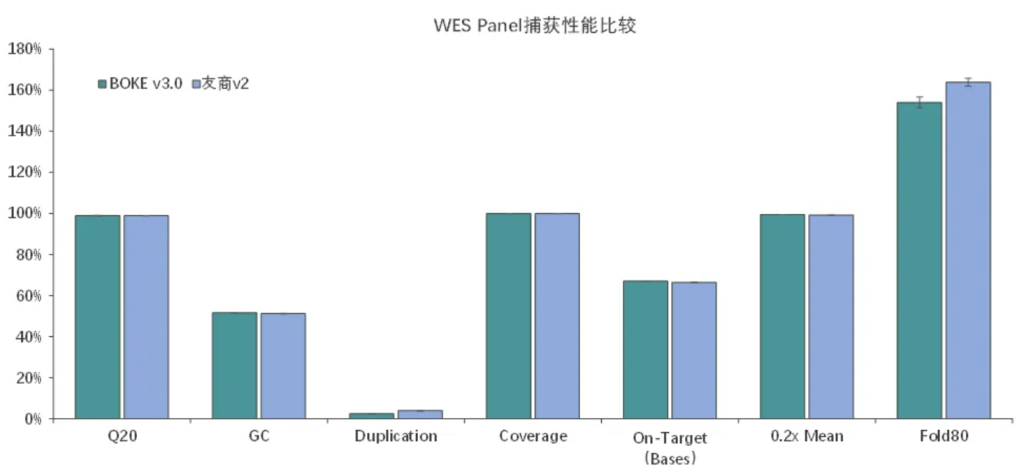
捕获性能比较
伯科全外芯片v3.0性能优异与国外友商同类型产品v2相当,中靶率、覆盖率、覆盖均一性等参数均达到国际领先水平。
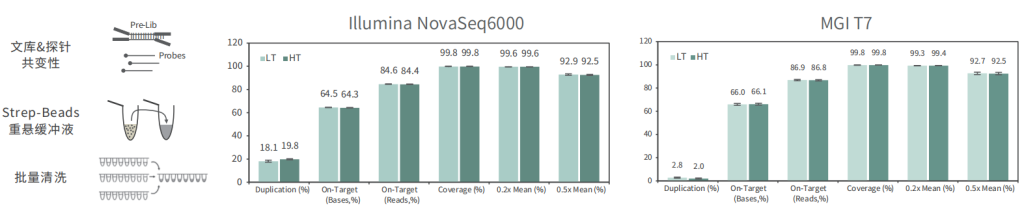
适配高通量流程平台
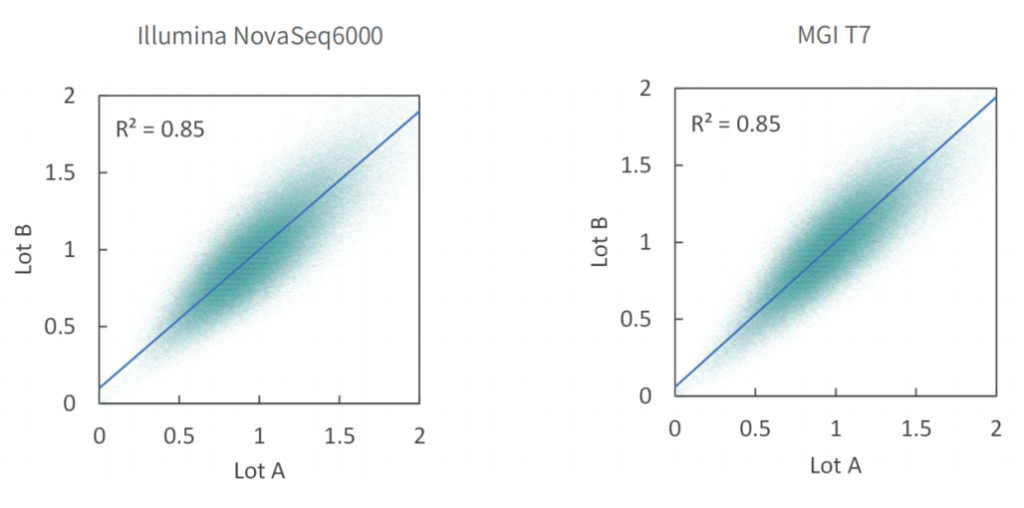
批次稳定
使用不同批次TargetCap® Core Exome Panel v3.0芯片对NA12878 gDNA进行捕获测序,结果显示,不同批次芯片在不同测序平台上均显示出优异的稳定性,不同位点的相对深度相关性高,批次稳定。
变异检测准确
单核苷酸变异(SNV)和插入缺失 (INDEL)是基因组变异的常见形式,也是引起人类疾病的重要原因。
选取NA12878标准品,与预期SNV和INDEL变异进行比较。结果表明,在MGI与Illumina测序平台,SNP灵敏度为99.1%,INDEL灵敏度为91.6%。
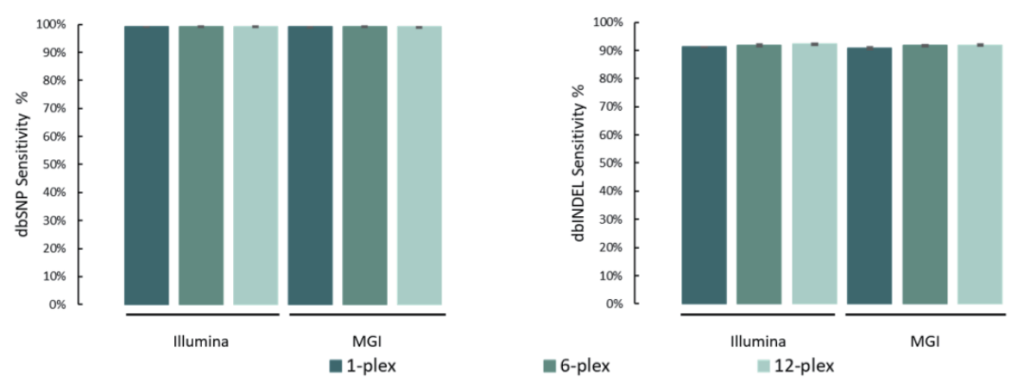
添加线粒体模块临床样本表现
20例全血样本(S1#-S20#),采用1-4 Plex方式使用TargetCap® Core Exome Panel v3.0添加线粒体模块进行过夜杂交捕获;其中,S1#-S10#在Illumina NovaSeq X Plus平台测序, S11#-S20#在MGI MGISEQ-T7平台测序,均采用150PE模式测序。得到测序数据后,抽取8Gb数据进行生信分析。
两种测序平台的数据表现相近,平均深度分别为111x/115x (Illumina/MGI),中靶率优异均> 85%,覆盖均一性极佳(0.2X_MD≥99.3%);仅使用8Gb数据,高达98.5%的捕获区域达到了30X以上,99.5%的捕获区域达到20X以上,为临床样本检测提供了可靠的捕获数据。
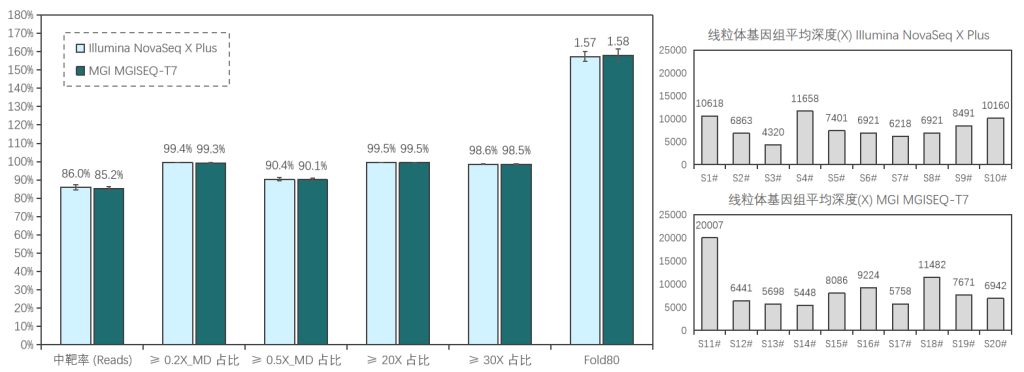
≥0.2X/0.5X_MD: Mean Depth,覆盖深度≥平均深度的0.2/0.5倍深度的区域占总区域的比例,用于表征覆盖均一性性能,越接近100%越好。
TargetCap® Core Exome Panel v7.0
TargetCap®Core Exome Panel v7.0(下文简称BOKE v7.0),该WES Panel增强了基因组hg19传统研究区域的覆盖,兼顾 hg19 & hg38 双版本基因组,可以更好的保证临床科研与转化的延续性。目标区域和捕获区域大小分别为40Mb和49Mb,对 hg19 传统研究区域覆盖提升至99.7%(友商I-v1),hg38传统研究区域覆盖相近(友商A-v8)。同时,新添加数百个具有一定功能与表型的基因,总基因数量达到20000+。
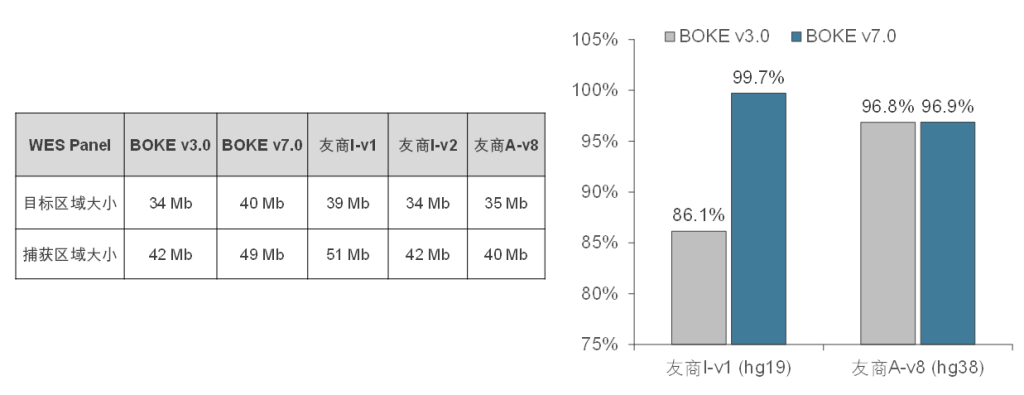
BOKE Core Exome Panel v7.0目标区域大小以及对不同友商产品目标区域的覆盖情况
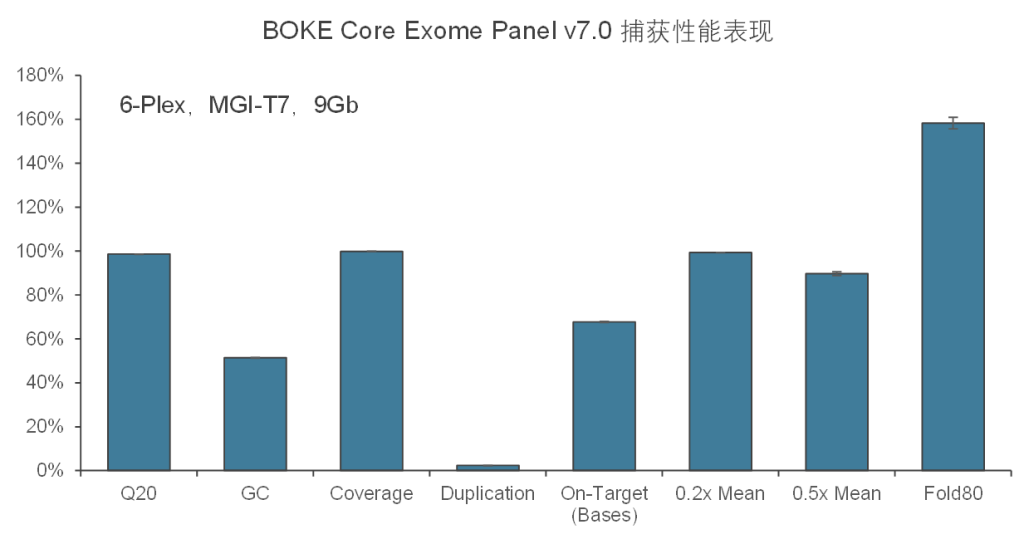
在捕获性能方面,TargetCap® Core Exome Panel v7.0依然表现优异,与TargetCap® Core Exome Panel v3.0表现相近。在测序9Gb条件下,平均深度达到110x左右,20x和30x以上区域占比分别为99.5%和98.5%,Fold 80为1.5-1.6之间,与国际领先产品数据表现相当。
同时,TargetCap® Core Exome Panel v7.0也可灵活的与拓展模块组合使用,满足不同场景的临床研究的需求及转化应用,包括线粒体、遗传病非编码区变异位点、单基因全覆盖、病毒基因组、肿瘤全景变异检测、重大疾病多基因风险评估模块等。此外,伯科公司自研自造的寡核苷酸合成平台可以快速响应个性化定制的需求,为人类基因组分子遗传学的研究与转化,提供更加全面高效的解决方案。
参考资料
Perry Elliott, Heribert Schunkert, Antoine Bondue, Elijah Behr, Lucie Carrier, Cornelia Van Duijn, Pablo García-Pavía, Pim van der Harst, Maryam Kavousi, Bart Loeys, Luis Rocha Lopes, Yigal Pinto, Alessandro Di Toro, Thomas Thum, Stefan Kääb, Mario Urtis, Eloisa Arbustini, Integration of genetic testing into diagnostic pathways for cardiomyopathies: a clinical consensus statement by the ESC Council on Cardiovascular Genomics, European Heart Journal, Volume 46, Issue 4, 21 January 2025, Pages 344–353, https://doi.org/10.1093/eurheartj/ehae747