


Genome Medicine | WES重分析全面解析先天性心脏病致病基因NODAL变异
- boke
- 2024-09-06
- 5:00 下午
心脏是脊椎动物胚胎中最早开始工作的器官之一。心脏发育始于原始心脏管的形成,这个过程发生在胚胎发育第三周结束时,伴随着胚胎的扭转折叠。原始心脏管形成后,需要打破左右(L-R)对称性,并经历一系列隔膜形成,最终形成四腔心。
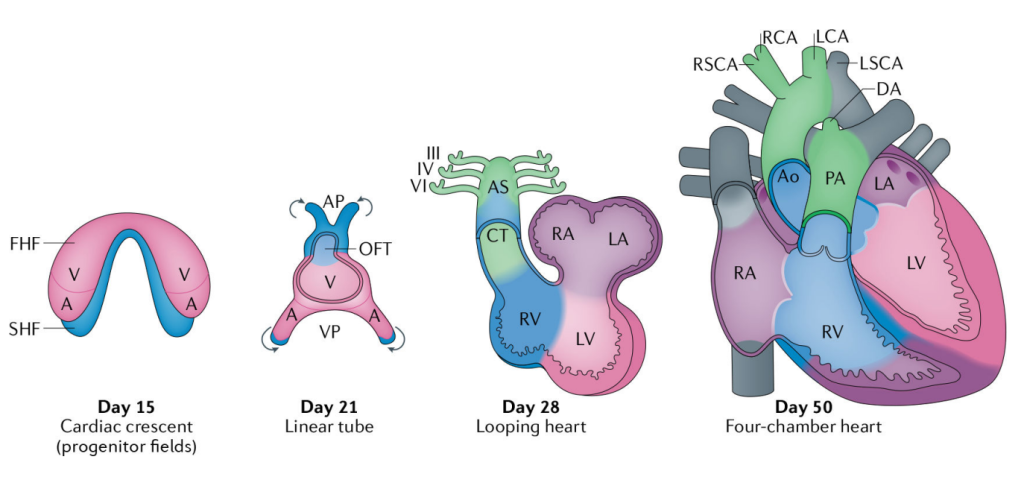
人类心脏发育过程[1]
心脏发育过程中细微的偏差会导致先天性心脏病(CHD),是胎儿最常见的先天性缺陷,所有新生儿的发生率约为0.8%[2,3]。
先天性心脏病的发生是遗传与环境因素共同作用的结果,很多患者并无家族史。目前已发现约1/3的先天性心脏病存在遗传学基础。CHD的病因很复杂,目前还不完全清楚。
先天性心脏病的遗传学基础主要包括染色体异常、拷贝数变异和基因变异。目前发现5%~20%的先天性心脏病患者存在致病性拷贝数变异,包括遗传和新发变异,其中拷贝数缺失较重复的后果更严重。单基因缺陷目前可以解释约 10% 的先天性心脏病,大多数致病/可能致病变异为新发变异。
很多CHD都跟左右不对称有关,左右不对称病因的遗传基础很复杂,我们对相关基因的了解还很有限,但常染色体显性、常染色体隐性和X连锁遗传模式在涉及异位的罕见病性状中均有观察到。
NODAL信号通路是左右轴发育的分子控制中的关键参与者,在脊椎动物胚胎和心脏发育中起着至关重要的作用。基因突变导致TGF-β/NODAL信号通路紊乱,已被证实会导致人类的内脏异位症。
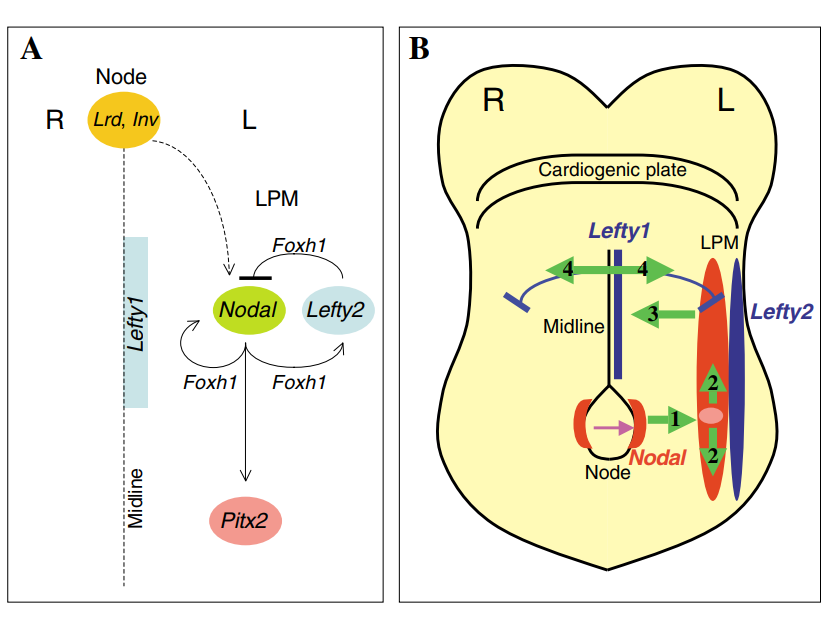
NODAL的left-right (LR) 模式调控关键组分[2]
在Genome Medicine最新发表的研究中[3],为了进一步探索这种关联并改善分子诊断,研究人员在一个大样本的左右不对称个体中,找到了更多不同的NODAL变异,然后详尽的研究了它们的可能致病机制。

方法
该研究使用基于家系的罕见变异基因组分析,重新分析了 321 名患有临床诊断的侧向性先天性心脏病 (CHD) 的先证者单样本全外显子测序数据,并向该队列添加了 12 名携带已知NODAL 变异的CHD 受试者,以调查等位基因类型。
Sanger 测序用于等位基因确认和遗传分离分析。Array-CGH和ddPCR 用于拷贝数变异 (CNV) 验证和表征。基于人类表型数据库的定量表型分析,以剖析等位基因特异性表型差异。
结果
在 33 例 CHD 病例中,NODAL 的错义、无义、剪接位点、插入缺失和/或结构变异被确定为异位和其他外侧性缺陷的潜在原因。
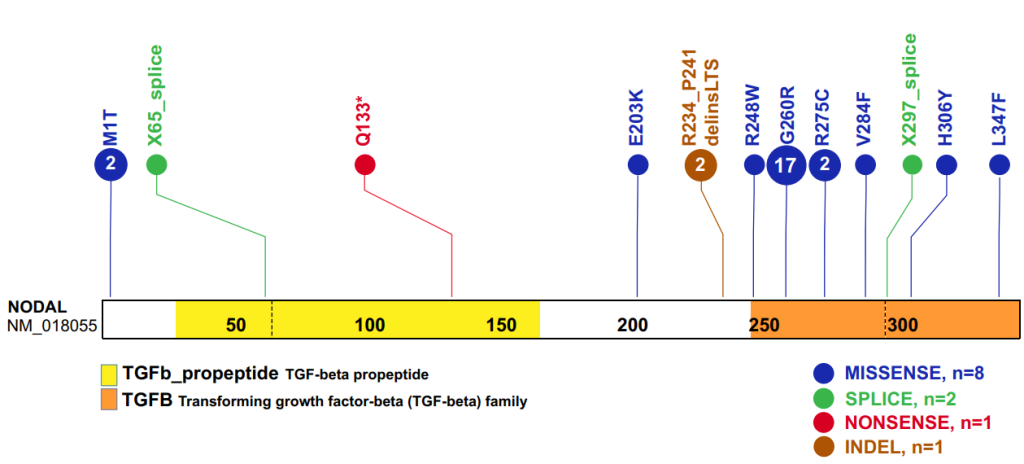
基于转录本 NM_018055 蛋白序列推导的 NODAL 蛋白结构示意图,图中显示了本研究中观察到的氨基酸变异的精确比对位置(垂直线)。我们发现 NODAL 结构域中共有 12 种不同的 SNV 等位基因和插入缺失。蓝色圆圈表示错义突变,绿色圆圈表示剪接位点突变,红色圆圈表示无义突变,棕色圆圈表示复杂的插入缺失突变。圆圈内的数字代表这些变异在研究队列中的频率。[3]
已鉴定的两个NODAL错义变异(c.2T>C,p.M1T;c.1039C>T,p.L347F)代表未报道的变异等位基因,以前从未与侧向性缺陷相关联。
在先证者(LAT0080)中鉴定的杂合p.M1T遗传给了他受影响的儿子(CVG005)。该变异与先证者及其儿子之间家庭内表型表达的可变性相关。先证者LAT0080表现为异位症、无脾、右心房异构、二尖瓣闭锁、室间隔缺损(VSD)、伴D型错位大动脉(D-MGA)的DORV和肺动脉狭窄(PS)。他还出现了严重的动静脉畸形(AVM)和膀胱输尿管反流(VUR);由于后者他接受了术后输尿管再植手术。而他的儿子(CVG005)表现为伴L型心室环的CCTGA、LTGA、肺动脉闭锁和VSD(表2和图3)。
病例LAT0048携带先前未报道的p.L347F变异,表现为DTGA。父母检测显示,这种罕见变异遗传自其没有CHD的父亲。347位亮氨酸是NODAL蛋白的最后一个NODAL 蛋白中的最后一个氨基酸,将其替换为苯丙氨酸预计会对 NODAL 功能产生有害影响,CADD 分数为 31,REVEL 分数为 0.51。
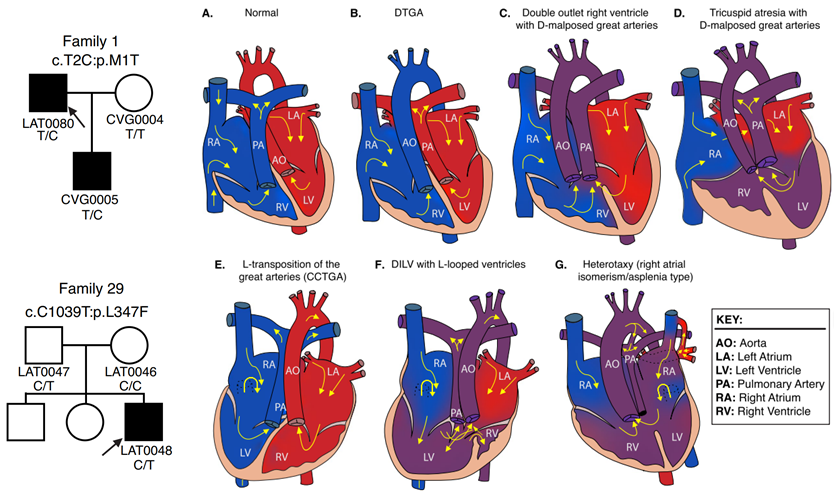
研究还发现了一种反复出现的复杂插入缺失变异,其DNA二级结构预测暗示二级结构诱变是一种可能的形成机制。
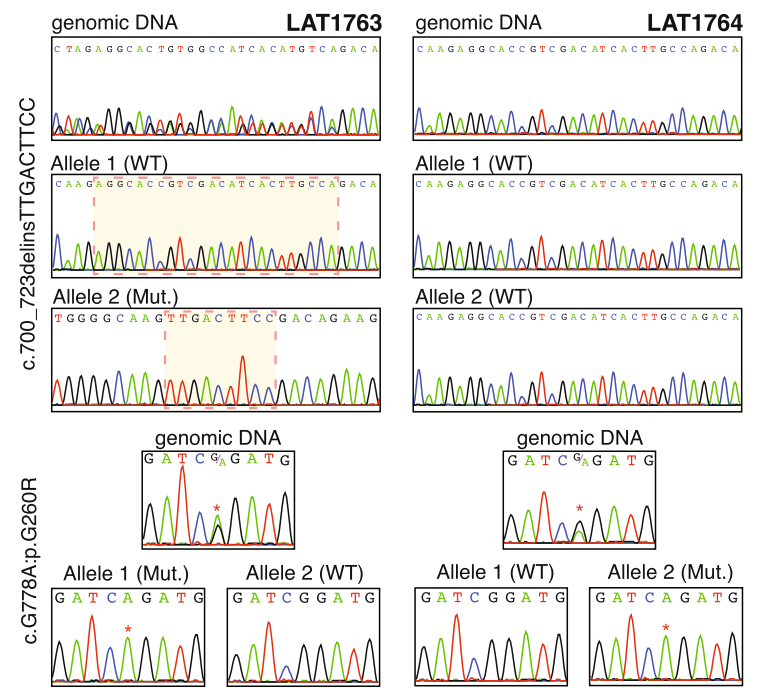
在两个无关的病例(LAT0191 和 LAT1763;分别是家系 6 和 7)中发现了一个插入缺失变异(p.R234_P241delinsLTS,c.700_723delins)。为了便于变异解释和验证,对先证者和父母的 DNA 进行了克隆实验,分离等位基因。然后进行了 Sanger 测序。这些实验证实,这两个先证者没有移码变异,但确实具有非移码 delinsLTS 变异等位基因。
需要注意的是,使用 RNAfold 服务器(http://rna.tbi.univie.ac.at/)对“野生型”(WT)和突变单链核酸进行的二级结构预测,应用最低能量和热力学系综函数进行分子内沃森-克里克碱基配对,表明这种复杂的突变是由二级结构诱变介导的。WT 结构的最低能量为 -10.40 kcal/mol,而突变结构的最低能量为 -13.20 kcal/mol(低 2.80 kcal/mol)。
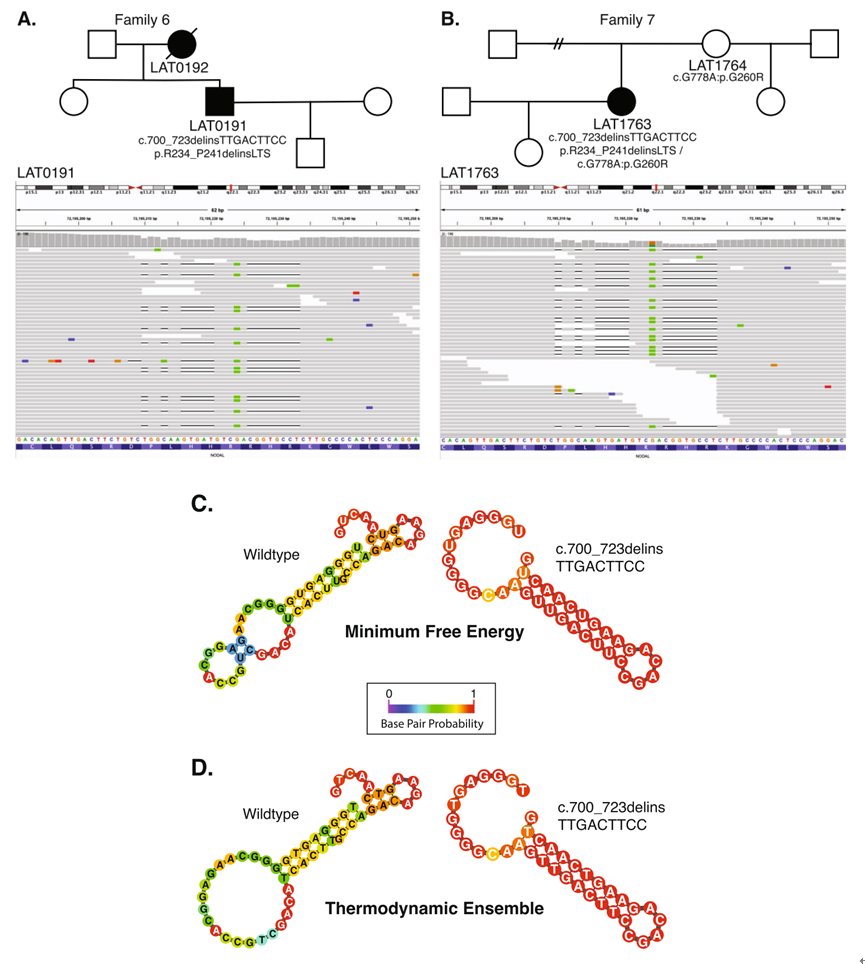
对于家系 7(LAT1763),除了 c.700_723delins 变异等位基因外,还检测到 c.778G>A:p.G260R 变异。LAT1763 的等位基因 1 在 c.700_723 位点是野生型,但包含 c.778G>A 变异。等位基因 2 包含 c.700_723delins 变异,但在 c.778G>A 位点是野生型,这表明这两个变异等位基因位于染色体上的不同位置,构成了复合杂合子。
该研究还在两个无关的 CHD 病例中发现了两个跨越 NODAL 的 CNV 缺失等位基因。此外,发现 17 名 CHD 个体(其中 16/17 名具有已知的西班牙裔血统)具有 c.778G>A:p.G260R NODAL 错义变异,因此建议将其从不确定意义的变异 (VUS) 重新分类为可能致病。对所有具有 p.G260R 变异的病例(包括杂合子、纯合子以及复合杂合子病例)的观察到的临床表型进行的基于 HPO 的定量分析,揭示了具有两个等位基因的个体的聚类。这一发现为基因型-表型相关性和等位基因特异性基因剂量效应提供了证据。
结论
总的来说,该研究结果表明,在异位病例中评估所有变异类型(单核苷酸变异、插入缺失和拷贝数变异)可以提高CHD病例的分子诊断率,并且等位基因特异性基因剂量可能是CHD外显率和变异表达的重要贡献者。该研究还发现NODAL中的罕见变异会导致异位谱系先天性心脏缺陷,并提供了证据表明,这些变异的群体差异应在该疾病的遗传咨询和临床基因组检测中予以考虑。此外,研究揭示了与侧向缺陷相关的未报道的NODAL突变和突变类型,这使我们能够进一步了解侧向缺陷背后的生物变化和遗传机制。这些发现对人类侧向缺陷的诊断和治疗具有重要意义。
参考资料
1. Morton SU, Quiat D, Seidman JG, Seidman CE. Genomic frontiers in congenital heart disease. Nat Rev Cardiol. 2022;19(1):26-42.
2. Shiratori H, Hamada H. The left-right axis in the mouse: from origin to morphology. Development. 2006;133(11):2095-2104.
3. Dardas Z, Fatih JM, Jolly A, et al. NODAL variants are associated with a continuum of laterality defects from simple D-transposition of the great arteries to heterotaxy. Genome Med. 2024;16(1):53. Published 2024 Apr 3.