


Genome Medicine | 应用全外显子测序(WES)揭示先天性肌病新致病基因并完善基因与表型的相关性
- boke
- 2024-08-29
- 3:55 下午

研究概述
背景
先天性肌病是严重的遗传性疾病,对患者的自主性有很大影响,并且常常与生存相关。大量患者未能获得遗传学诊断,这阻碍了遗传咨询和适当的临床管理。研究目标在于发现与先天性肌病相关的新致病变异和基因,以缩短漫长的诊断旅程并避免无果而终。
方法
为了确定先天性肌病的致病变异和致病基因,在 2009年至2018年期间建立并开展了 MYOCAPTURE 项目,对310个部分排除了主要已知基因的家系进行了全外显子组测序。
结果
在156个家系中(占50%)发现了致病变异,其中123个家系(占40%)获得了明确的诊断。在这些确诊病例中,仅有44例(占36%)与已知的肌病基因及其相应表型相符,而55例(占44%)则与已知肌病基因中的致病变异相关,但表现出非典型症状,这表明在该群体中,大多数遗传诊断无法仅凭临床-组织学评估判断。不同基因之间以及不同先天性肌病亚型之间均观察到了显著的表型和遗传异质性。此外,还发现了14个以前未在文献中报道过的与肌肉疾病相关的新肌病基因(占所有确诊病例的 20%),揭示了新的病理机制和潜在的治疗靶点。
结论
总的来说,该研究展示了WES高通量基因测序作为一项全面工具,在为患有先天性肌病的家系建立分子诊断方面的重要性。同时,也强调了临床数据、肌肉活检的组织学发现以及来自其他家系成员的DNA样本的可用性对提高诊断成功率的贡献。这项研究促进了先天性肌病的遗传诊断,加速了诊断过程,改善了多名患者的医疗护理,并为现有分子的再利用或新治疗方法的开发开辟了新的视角。
研究背景
先天性肌病(CM)是一组罕见且严重的遗传性疾病,对患者的自主能力和生存状况产生深远影响。迄今为止,许多患者仍未获得遗传学诊断,这阻碍了更有效的医疗护理、潜在治疗手段的应用以及包括产前诊断在内的遗传咨询的实施。
本研究旨在缩短诊断过程中的迷茫与困境,并发现新的致病基因,这些基因或将成为新的治疗靶点。
先天性肌病与肌肉无力和/或低肌张力有关,通常在出生时或婴儿早期出现,成年发病的情况较少。此外,心肌病、骨骼畸形或呼吸衰竭等其他症状也经常伴随出现。
先天性肌病与肌肉坏死和再生无关,因此不同于肌营养不良症。相反,先天性肌病患者的肌肉活检通常显示出特定亚型的结构异常。
在过去十年中,导致先天性肌病的基因数量从19个增加到47个,其中大多数编码肌丝或调节钙耦合肌肉收缩的蛋白质。
2012年之前,基因诊断通常是基于临床和组织病理学发现,逐一针对特定基因进行,这一过程受限于对相关基因知识的了解,有时还会因某些基因的过大(如TTN、RYRI、NEB基因)而受阻。
到了2012年,针对已知神经肌肉疾病相关基因的部分或全部区域的Gene Panel技术得到了验证。如今,在常规诊断实验室中,对于那些通过目标PCR(例如DMPK基因)或酶活性检测排除常见遗传变异的患者,通常会采用下一代测序技术(NGS)进行全外显子(WES)和全基因组(WGS)测序。
在此项研究中,为了识别与先天性肌无力(CM)相关的有害变异和基因,组建了一个由310个家系组成的大型队列,这些家系根据临床和组织病理学发现被分类为同质的CM亚型。通过对先证者及其亲属的DNA样本进行WES测序和内部生物信息学工具分析,并结合遗传学和功能性研究来确认候选变异/基因。从310个家系中收集的DNA样本,涵盖了429名患者和459名未受影响的亲属。
在WES数据分析和变异识别中,根据家系结构(隐性纯合/复合杂合、显性、新生、X连锁及其他特定情景)应用分离情景。变异过滤基于gnomAD数据库和内部NGS数据库中的群体频率。随后,使用VaRank算法对变异进行加权和优先级排序。对于错义变异,采用SIFT或PolyPhen进行体内致病性预测。对于剪接变异,使用MaxEntScan、NNSplice等剪接变异预测工具。
对于一部分同义、典型或非典型剪接位点的变异体,采用从肌肉中提取的RNA的RT-PCR产物进行cDNA测序,以证实其对外显子剪接的致病性影响。对于某些变异体,需要进行分离研究以确定其致病性。在缺乏亲属DNA样本的情况下,这些变异体被归类为候选变异。变异体的分类遵循ACMG标准和指南。
部分研究结果
自2009年至2018年间,本研究共纳入了310个家系,这些家系的先天性肌无力症(CM)尚未通过遗传学手段确诊。在214个家系中,至少有一名亲属可供研究,而在92个家系中,则包括了家系本人及其双亲在内的完整三口之家。
总计,通过WES测序或Sanger测序法,对429名个体进行了遗传分离研究,大多数患者为散发病例(占78%)。在25个家系中,父母双方被报告为近亲结婚。
如图1A所示,175名先证者来自法国(占总数的57%),102名先证者来自15个国家(占总数的32%),另有32名先证者的来源国未知(占总数的11%)。
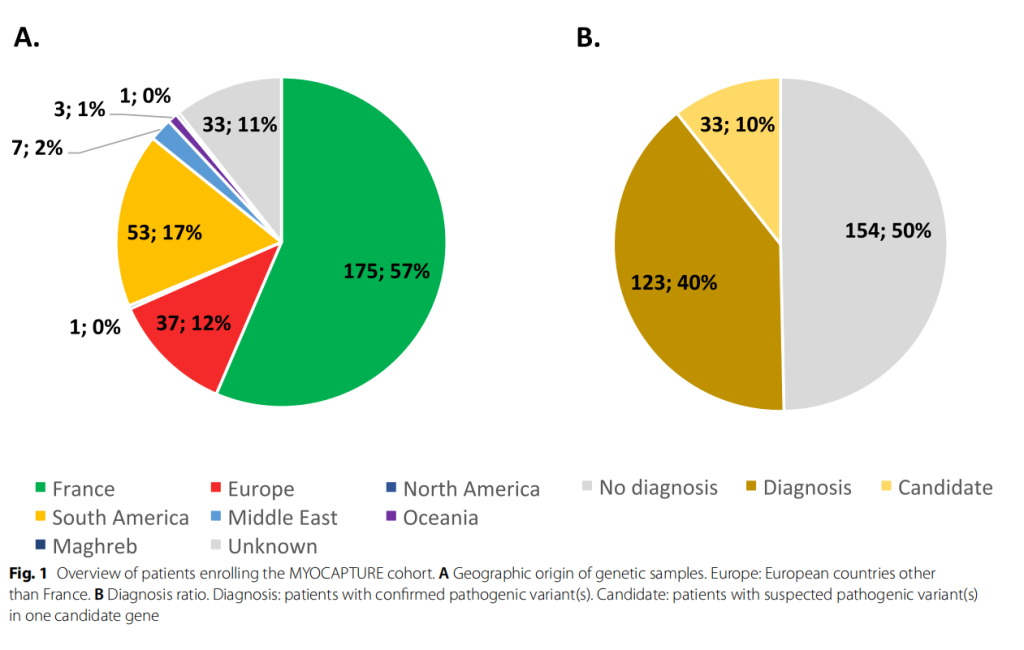
图1. MYOCAPTURE 队列患者概览。
家系、肌肉活检的重要性
WES测序确定了123个家系(40%)的致病变异(图 1B)。在33个家系(占10%)中,检测到了在有说服力的候选基因中的序列变异,但由于缺乏亲属的DNA样本,无法进行遗传分离分析。
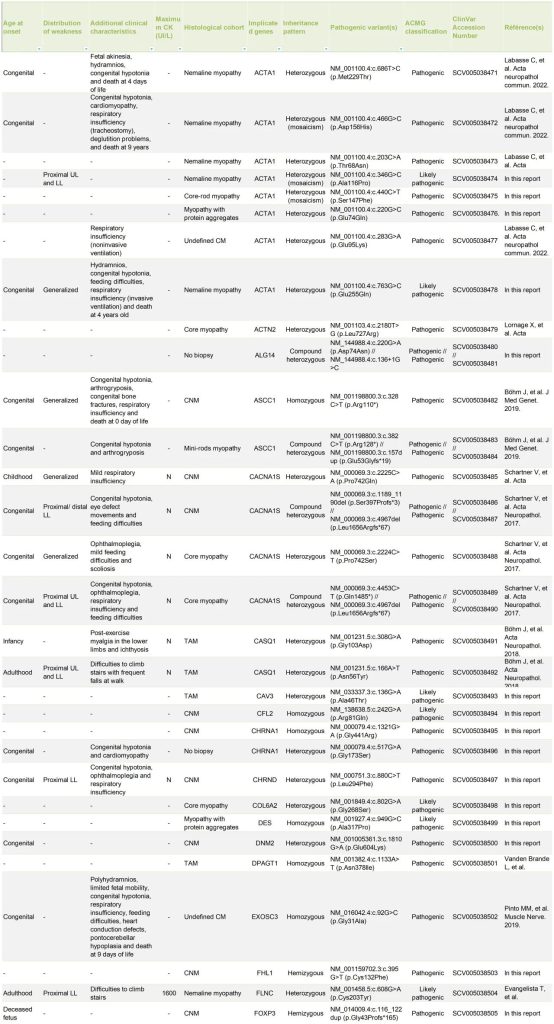
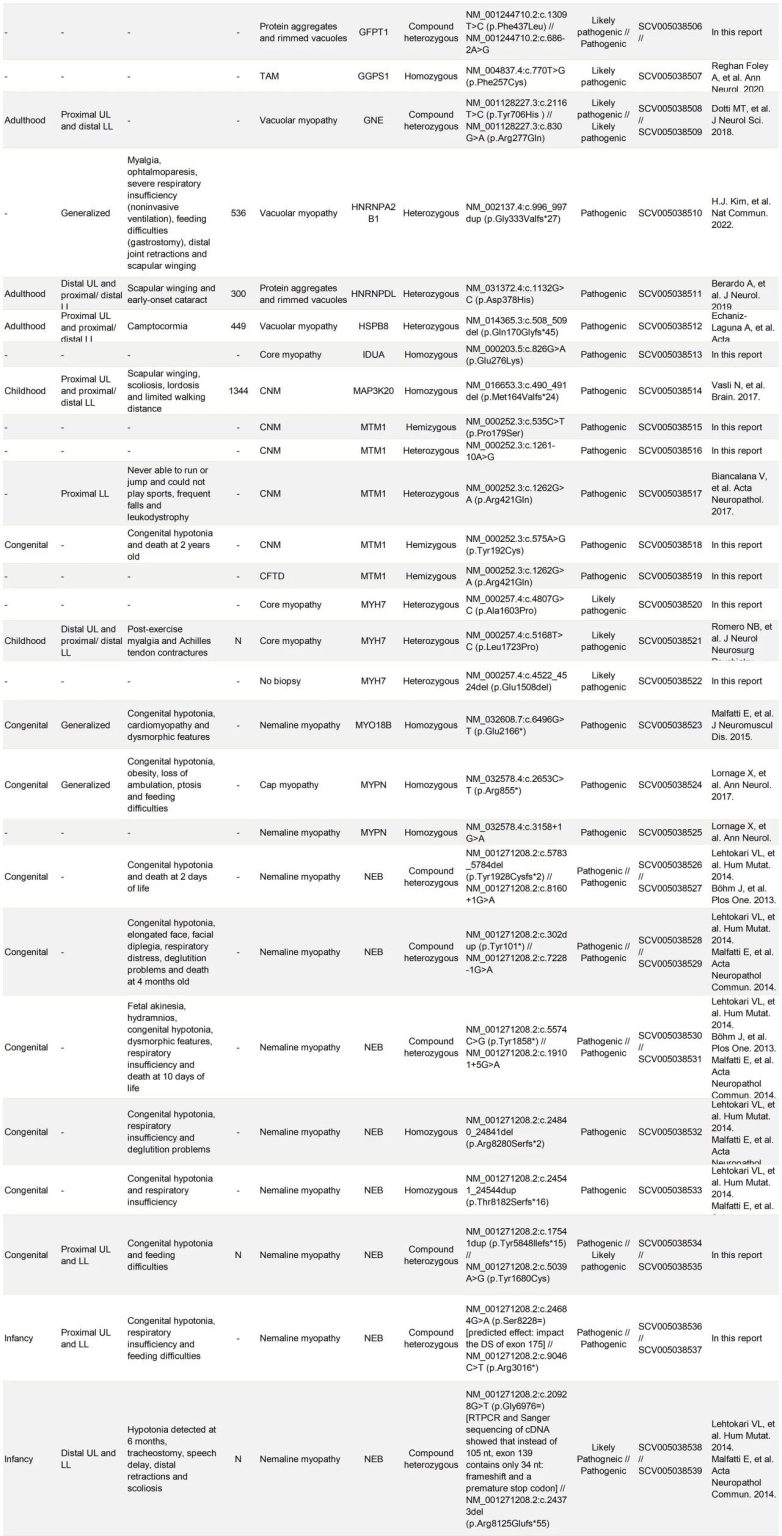
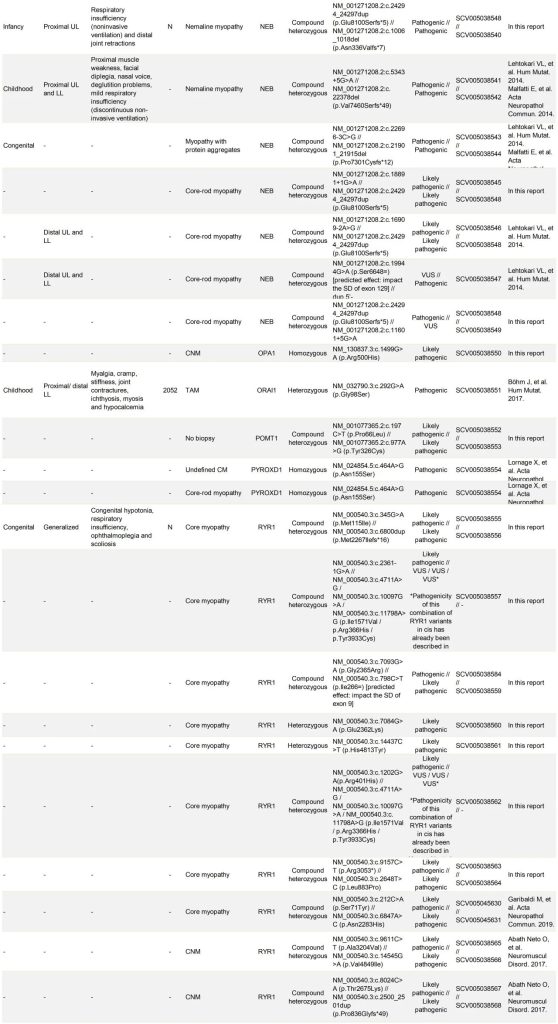
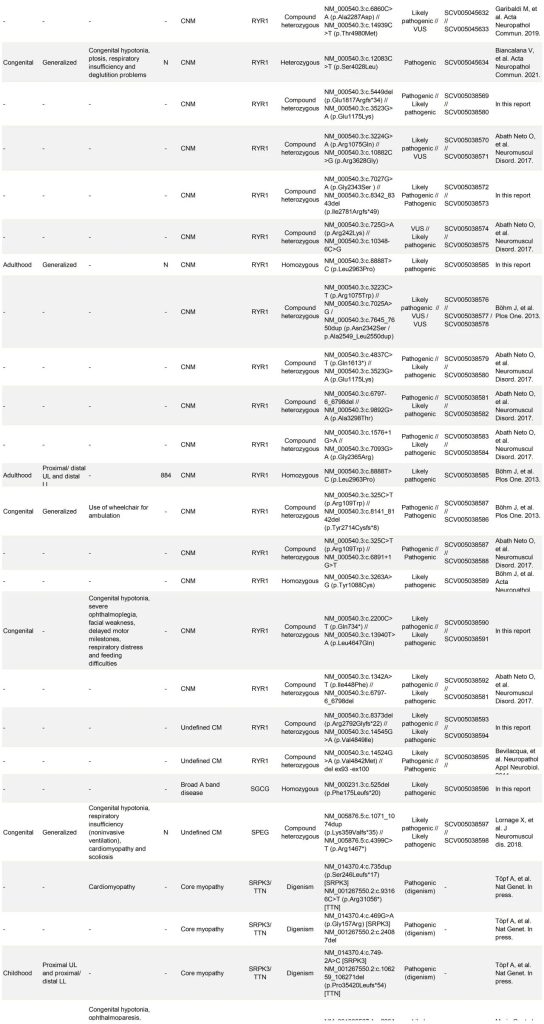
表S1. 123个家系中先证者的临床、组织学和致病性变异信息
在该研究群体中,测序的家系成员数量与诊断率之间存在相关性。增加测序的家系成员数量提高了致病变异识别的可能性,其中大多数家系(51%)在至少有三个DNA样本可用时被诊断出来(表S1)。与双人组(29%)相比,三人组的诊断家系比例更高(48%)。
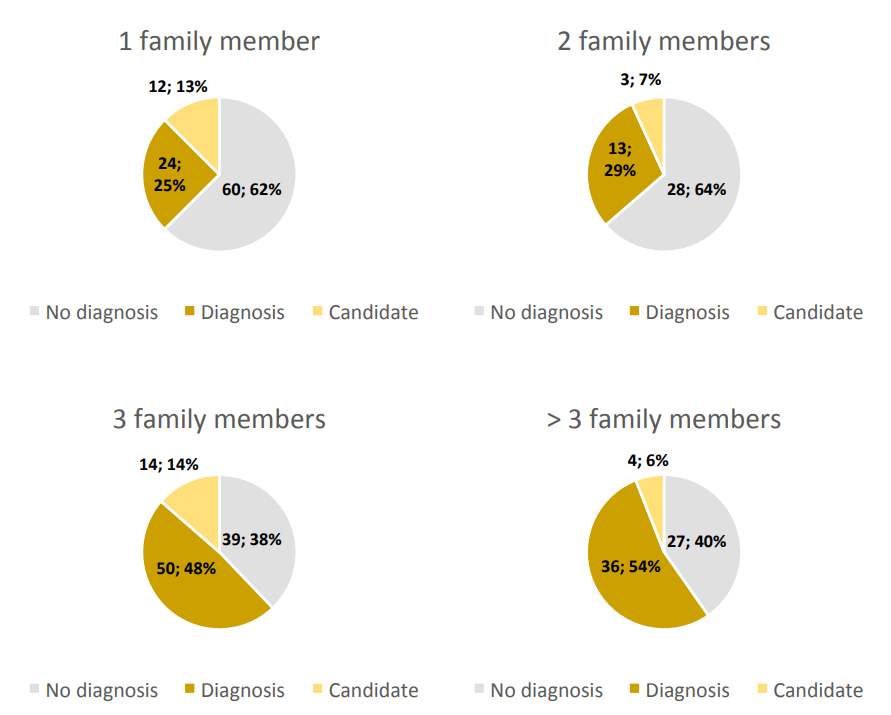
图S1. 家系可用DNA数量与诊断率的关系
同样,随着每个家系中受影响个体数量的增加,识别出致病变异的家系比例也增加,大多数家系(55%)在至少有两个受影响个体时被诊断出来(图S2)。
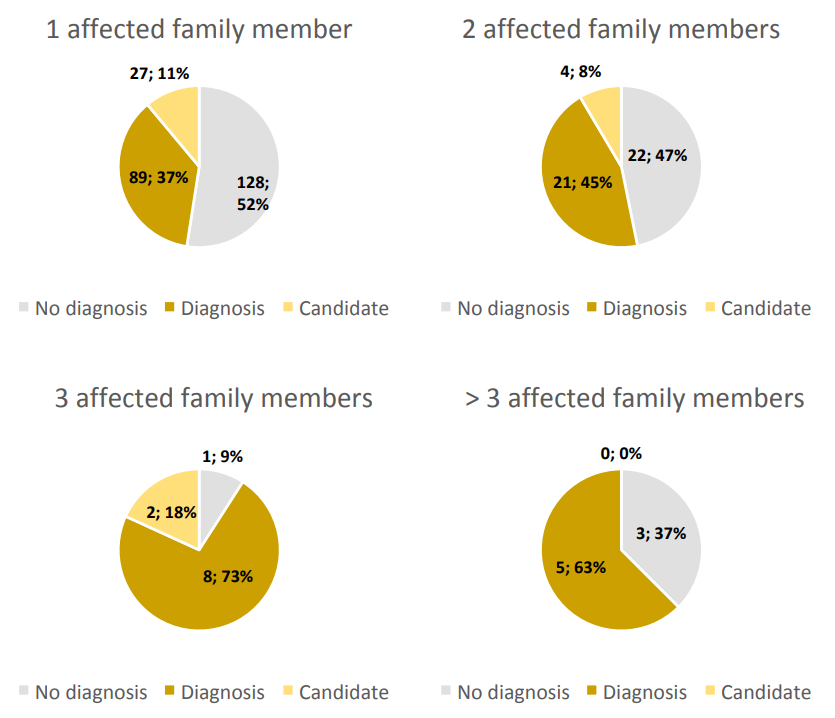
图S2. 家系受影响个体与诊断率的关系
通过组织学检查的肌肉活检是另一个对分子诊断有重要贡献的因素。实际上,在有活检的家系中,42%的家系发现了致病性变异,而在没有活检的家系中,这一比例仅为14%(图S3)。这可能部分是因为临床表型最严重的患者更常进行活检;然而,这些结果支持除了临床评估之外进行补充性调查的重要性,例如组织病理学,以准确诊断CM并优先考虑候选基因。
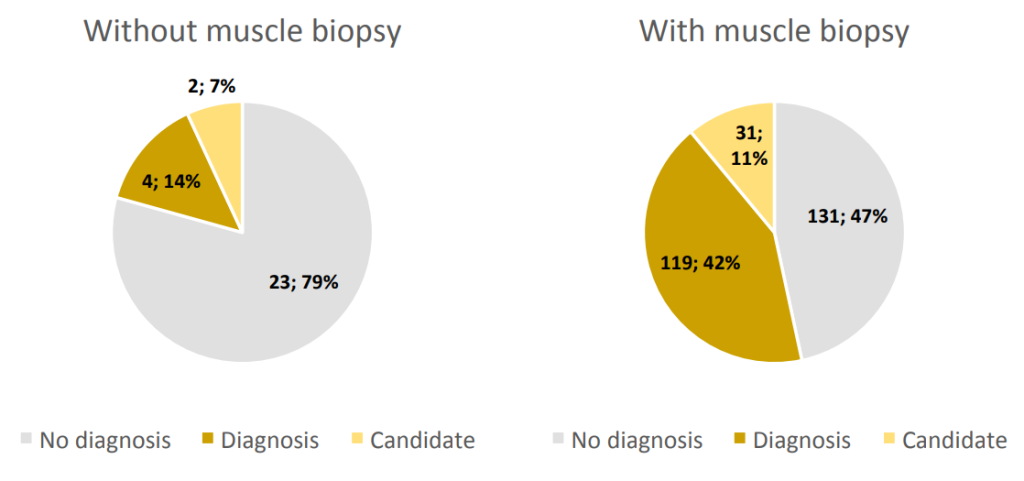
非编码区域和完整基因分析的重要性
在经过分子诊断确认的123个家系中,发现了47个不同的致病基因。值得注意的是,有4个基因在这些家系中出现的频率异常高,它们在60个家系中(占总数的49%)与疾病相关的变异体特别多。具体来说,RYRI基因涉及的病例最多(共29例,占24%),其次是NEB基因(15例,占12%),以及TTN和ACTA1基因(各8例,占7%)。
在123个确诊的家系中,有16个家系(占13%)携带了可能容易逃过常规诊断的致病变异。在MTM1(1例)、RYR1(1例)、UNC4SB(1例)和NEB(4例)的非典型剪接位点中发现了7种致病变异。
在三个患有NM的家系、一个核心肌病家系和一个CNM家系中,分别发现了五种影响剪接的NEB、RYR1和TTN同义变异。
三个患有NM的家系中,ACTA1基因存在可在肌肉DNA检测到的嵌合致病变异,血液DNA勉强可检测到。
最后,在两个表现出症状的CNM女性携带者中检测到了杂合的MTM致病变异,而在一个CNM家系中,先前被认为是多态性的一个RYR1错义变异现在已被重新归类为致病变异。总的来说,这些例子强调了彻底检查非编码区域的重要性,并使用预测程序来评估的重要性。
在确诊的123个家系中,有11个家系(占9%)存在致病变异,这些变异未被检测到的原因是这些家系中存在RYR1基因(共8例)的致病变异,该基因先前仅针对最3’末端的外显子进行测序;或者是ACTA1基因(共3例)中的嵌合型致病变异,这些变异在常规的血液DNA遗传检测中未被发现。
讨论
该研究结果证实了全外显子测序(WES)在为患有未确诊先天性肌病的患者提供遗传诊断方面的效率,尤其是在结合临床和组织病理学数据,以及获取额外家系成员信息的情况下。
该研究项目主要基于对310个遗传诊断未明的CM家系进行的全外显子测序(WES),这些家系根据临床和组织病理学发现被分类为同质群体。在50%的家系中发现了致病变异,其中79%的家系获得了明确诊断。
对于剩余的21%,在肌肉相关基因中识别出了可能具有致病效应的序列变异。值得注意的是,仅有36%的已确诊患者与已知的肌病基因及其一致的表型相关联,这表明在大多数情况下,临床和组织学评估不足以预测分子诊断,这强调了大多数遗传诊断无法通过仅限于临床-组织病理学评估的已知基因测序找到。这包括了先前与其它肌病、神经肌肉疾病或其他综合征相关联的基因。
此外,发现了14个新的肌病相关基因,揭示了新的病理机制和潜在的治疗靶点。总体而言,这些结果促进了未解决家系的遗传诊断进程,加速了诊断速度,改善了多名患者的医疗护理,并为现有分子的重利用或新治疗方法的开发开辟了新的方向。