


Genome Biology 解析人类最复杂基因的“暗区” | 迄今为止,LPA基因 KIV-2 序列变异最大规模分析
- boke
- 2024-08-28
- 5:23 下午
不同维度的研究均表明,脂蛋白(a)[Lp(a)]是心脑血管疾病(CVD)的致病风险因素,与低密度脂蛋白(LDL)不同,Lp(a)水平不能通过生活方式干预。不过有些个体天然具有较低的Lp(a)水平,他们的CVD发病风险更低,其内因在于Lp(a)的遗传学特点。
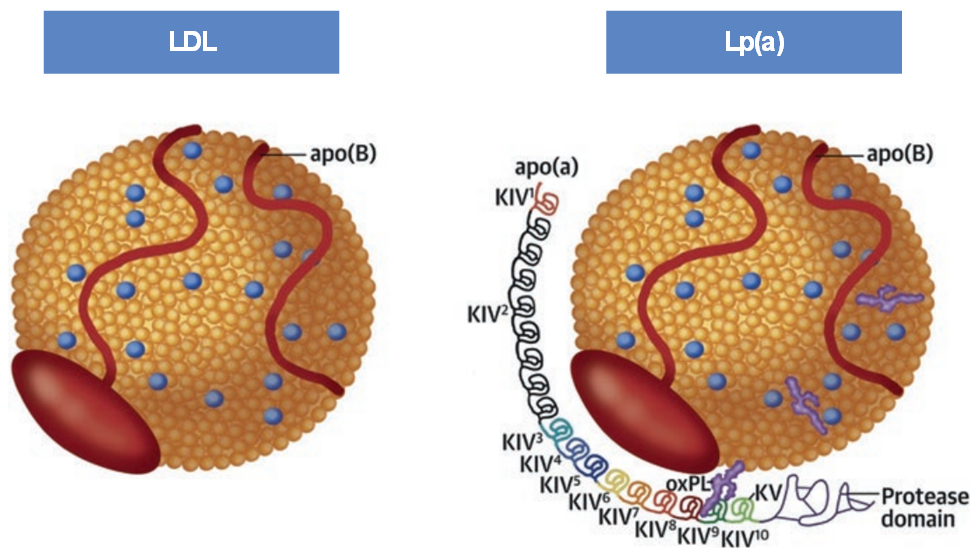
Lp(a)的蛋白质结构由两种载脂蛋白,ApoB-100和Apo(a)组成,两者通过单一的二硫键共价结合,Lp(a)水平高低主要由其组分中Apo(a)的编码基因-LPA决定。LPA基因的特征主要包括VNTR拷贝数和序列变异两个方面,两者共同决定了Lp(a)的浓度水平。
LPA基因与PLG基因高度同源,在LPA基因中存在一种叫作VNTR(可变串联重复)的区域(KIV-2),这个区域是由~5.5Kb作为基数长度的片段串联而成,不同个体的串联片段数量不等,范围约为1~ >40个拷贝。此外,人类基因组是二倍体,所以这种组合理论上达到了惊人的~1600种可能,具有极高的多态性。目前的研究表明,KIV-2拷贝数与Lp(a)水平负相关,KIV-2拷贝数越少,Lp(a)水平越高。
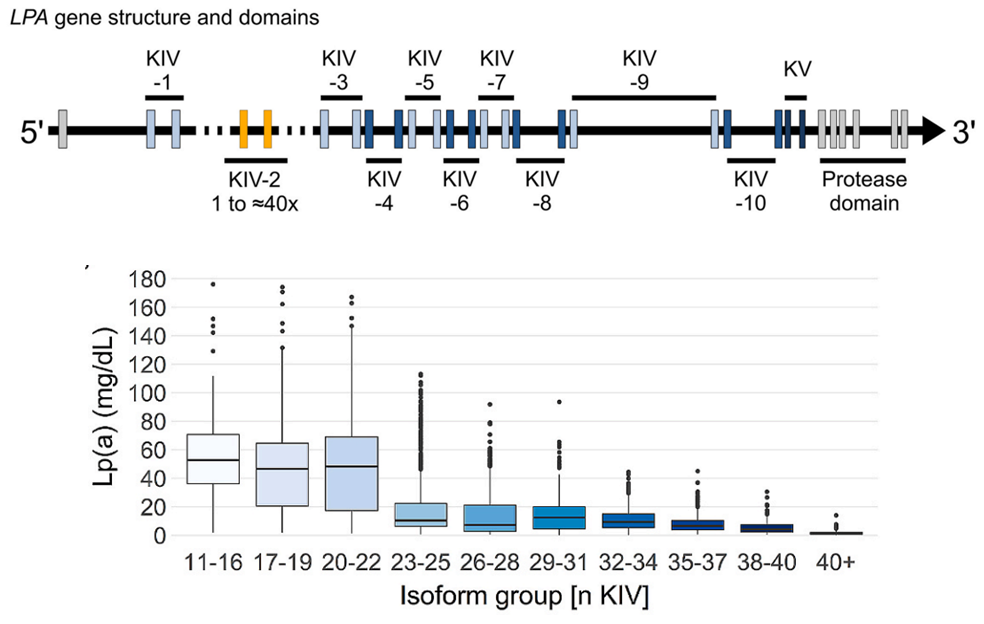
图2. LPA基因结构(上),Lp(a)浓度与KIV-2拷贝数负相关(下;n KIV表示KIV-2在内的KIV拷贝总数)[2]
同时,在LPA基因中还存在许多序列变异(功能性位点),这些变异往往可以降低Lp(a)水平,它们也被称为“保护性变异”。换言之,即使具有低拷贝KIV-2,这些个体的Lp(a)也可以保持在较低水平,并具有更低的CVD风险。表1显示了23个重要的功能性位点,除rs1800769外,其他位点的功能均是降低Lp(a)水平,值得注意的是,约30%的功能位点位于KIV-2区域内部。

表1. LPA基因的KIV-2拷贝数与23个功能性位点变异可以解释~90%欧美人群Lp(a)水平[3]
功能性变异如此高频的发生在KIV-2区域,可能与KIV-2本身是VNTR区域,且包含了LPA高达70%的编码蛋白质的序列有关。然而,尽管KIV-2区域具有许多潜在的功能性变异,由于VNTR的重复性,KIV-2区域内部的序列变异检测仍然面临具较大挑战,科学们也将LPA基因的KIV-2称为悬而未决的“暗区域”。
近期,发表在Genome Biology期刊上的一项研究针对LPA基因KlV-2 VNTR开发了一种优化的检测方法,可预先分辨出混合的KlV-2亚型。与先前的研究策略相比,该方法在F1得分上实现了2.1倍的提升。对近20万例英国生物样本库(UK Biobank)进行分析,检测到了超过700种KlV-2突变。这一方法不仅成功揭示了KlV-2变异体对降低Lp(a)水平的新的强效作用,并发现其对冠心病具有保护功能,同时还验证了先前研究结果中基于SNPs的一些标记Marker[4]。
该方法不仅为LPA基因KIV-2变异检测铺平了道路,同样也可应用于其他基因的VNTR检测,包括NEB(多种肌病有关,具有人类最长的VNTR重复单元>10Kb)、DMBT1(家族性胶质瘤)、FLG(特应性皮炎)、SPDYE3(屈光不正)和UBC(高密度脂蛋白胆固醇)五个与医学相关的VNTR基因,这将有助于揭示隐藏在VNTR中的医学信息。

背景介绍
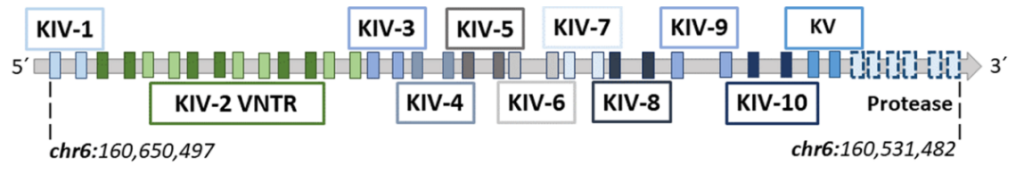
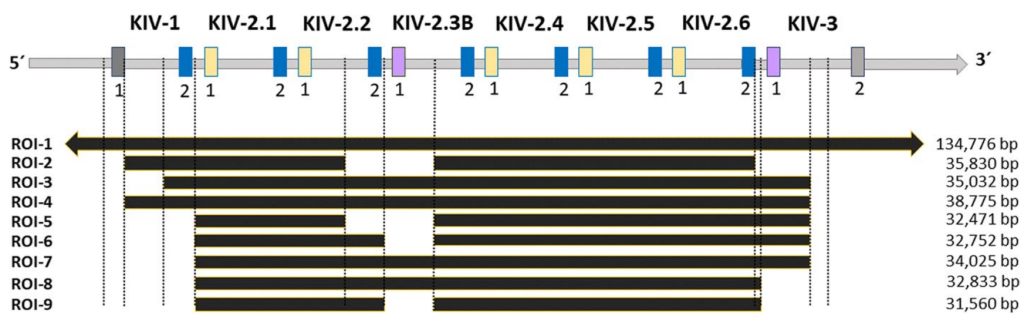
LPA包含10个高度同源的kringle IV (KIV)结构域(图3),其中KIV-2结构域是一个VNTR,每个等位基因有多达40次重复,它可以涵盖高达LPA编码序列的70%。
每个KIV-2单元大约为~5.5 kb大,形成一个大约200 kb大的VNTR数组,一个仅存在于一个重复中的变异可能仅显示出1/80=1.25%的突变频率。
每个KIV结构域由两个短外显子编码(大多为160 bp和182 bp),外显子之间间隔大约为4 kb的内含子,一个~1.2 kb的内含子连接单个KIV结构域(内宽外窄)。
KIV-2的外显子与邻近的KIV单位(尤其是KIV-1和KIV-3)的相应外显子高达98%的碱基同源性。如图4所示,KIV-1的第二个外显子与KIV-2的第二个外显子相同(蓝色),KIV-3的第一个外显子与KIV-2.3B第一个外显子相同(紫色)。我们还注意到,KIV-2的第一个外显子还存在黄色的类型,这与KIV-2的亚型分类有关
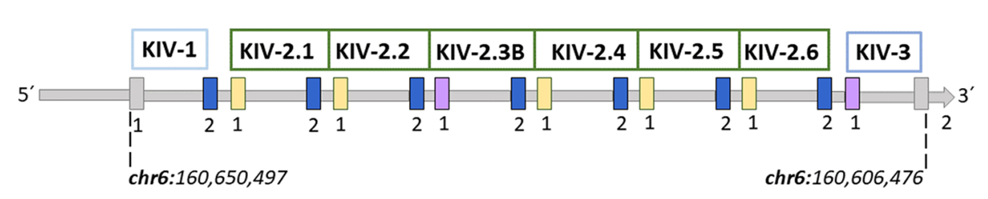
图4. 参考基因组中,KIV-1、KIV-2与KIV-3结构域细节。
实际上,KIV-2结构域至少存在三种几乎相同的亚型(称为KIV-2A、B和C),它们按照KIV-2第一个外显子的序列差异进行分类(图5)。
目前人类基因组参考序列中,KIV-2包括6个重复单元,KIV-2.1~KIV-2.6。由于KIV-2C型较为罕见,因此基因组中只包括了两种KIV-2亚型,KIV-2A(图4. 黄+蓝:5拷贝,KIV-2.1~2/4~6)和KIV-2B(图4. 紫+蓝:1拷贝,KIV-2.3B),其中,KIV-2B亚型对应的KIV-2.3单元是作者为了便于描述,标记为KIV-2.3B。
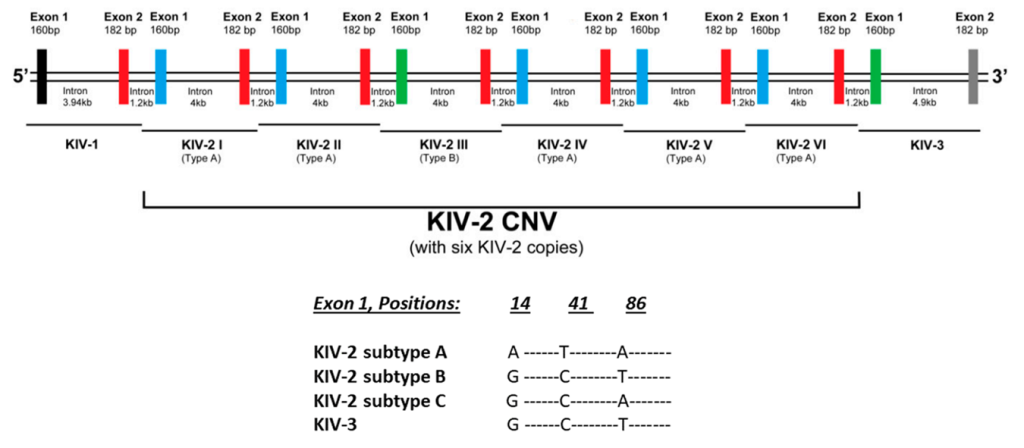
图5. KIV-2亚型分类根据以及其在参考基因组分布中的分布,红色外显子与图4蓝色外显子对应,天蓝色外显子与图4黄色外显子对应,绿色外显子与图4紫色外显子对应[Schmidt K, Noureen A, Kronenberg F, Utermann G. Structure, function, and genetics of lipoprotein (a). J Lipid Res. 2016;57(8):1339-1359.]
如图6所示,KIV-2内部、KIV-2与KIV之间的序列高度同源性,使得VNTR边界的定义、序列比对和突变检测极为复杂,KIV之间的错位比对将产生大量的假阳性Calling(旁系同源序列变异-Paralogous Sequence Variants, PSVs)。
1)KIV-2结构域的三种亚型(KIV-2A、B和C)几乎相同,它们之间的区别存在三个同义变异差异以及>100个内含子变异;
2)KIV-1的第二个外显子与KIV-2的第二个外显子相同,甚至在外显子周围的前200个内含子碱基中也观察到大约70%的同源性。KIV-2B的第一个外显子与KIV-3的第一个外显子相同,而相应的第二个外显子显示出96%的碱基同一性;
3)其他高度同源的KIV单位( 如 KIV-1、KIV-3、KIV-4 )。

图6. KIV Domain部分序列比对情况
上述复杂性阻碍了LPA中的突变筛选,因此在现有的变异数据库中,LPA仍未得到有效解决,这也导致其被排除在最新关于复杂基因组区域变异的基准数据集之外。
为了绕过这些难题,研究者先前采用了针对KIV-2重复序列的扩增子测序,这种扩增子以高度特异性的方式仅扩增KIV-2单元[2](图7A)。经过深度测序后,所有来自这些扩增子的读取序列都被比对到一个KIV-2重复序列上,并将KIV-2突变识别为低频突变,排除了来自同源区域的干扰读取。通过这种扩增子测序的方法,发现了KIV-2区域具有高度的变异性,而且即使是高度频繁的功能性变异,也长期未被发现,例如4925G > A携带者频率为~22%,4733G>A为38%。
然而,目前的基因组数据多以靶向捕获(WES、Gene Panel等)和全基因组测序(WGS)为主,由于LPA基因内部及KIV-2亚型之间广泛的同源性,这给从WGS或靶向捕获数据中对KIV-2 VNTR进行变异识别带来了额外的复杂性,因为这些数据包含了来自所有KIV单元的读取信息,因此,它们特别容易受到由来自同源KIV单元和/或不同KIV-2亚型的错误比对所引起的假阳性变异识别的影响(图7B)。
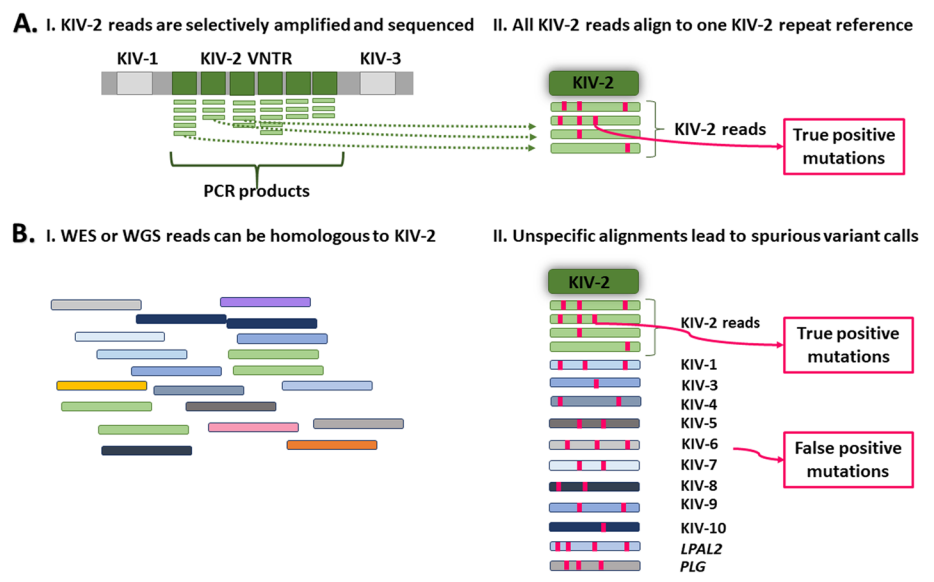
图7.扩增子测序与靶向捕获技术测序、全基因组测序对KIV-2变异检测的差异
该研究利用高度特异性的基于扩增子测序的数据集作为“金标准”,用于评估并改进从WGS或WES数据中对LPA KIV-2 VNTR的变异识别。为此,他们建立了一种通过特定PSVs开展样本个体化的动态优化方式进行变异检测策略,而提高LPA KIV-2变异识别的准确性。最终,将这一策略应用于来自UK Biobank的超过199,000份WES样本,完成了迄今为止对LPA KIV-2 VNTR最大规模的分析。
研究结果
研究者提出了一种计算方法,用于解析从短读序数据中获得VNTRs内的碱基变异,特别是针对LPA基因中的KIV-2 VNTR,该区域因其两侧存在高度同源域而成为一个特别难以处理的目标。
借鉴先前在LPA基因研究成果,提供了一个通用且便于使用的分析流程,用于在VNTR重复序列中检测碱基变异,并专门解析复杂的LPA KIV-2 VNTR内的变异。
该工作流程能够分离出目标VNTR的Reads,并且无需调整即可应用于来自千人基因组计划(1kGP)的2504个样本中的其他5个蛋白质编码VNTRs(NEB、DMBT1、FLG、SPDYE3和LUBC)。
在LPA基因的实验中发现,Reads提取策略的差异会显著影响变异检测的性能,更严格的坐标定位能带来最准确的结果。此外,该研究还展示了如何利用侧翼非重复区域的单核苷酸多态性(SNPs)来个体化地识别KIV-2 VNTR亚型,并动态调整VNTR Reads的识别。
一、VNTRs 变异检测生物信息分析流程
生物信息分析流程如图8所示,输入(Input)是所有WES/WGS Reads预先比对到人类参考基因组的BAM格式文件。所有比对到用户自定义的VNTR ROI(用户感兴趣的VNTR区域)的Reads都会被提取并重新比对到由单个重复单元组成的参考序列中。单个VNTR单元中的变异仅出现在部分Reads中,类似于体细胞突变,因此使用mutserve变异检测器在优化的低频变异检测设置下进行调用。为了简化和扩展到用户定义的VNTR的应用,该流程使用Nextflow工作流管理器实现。
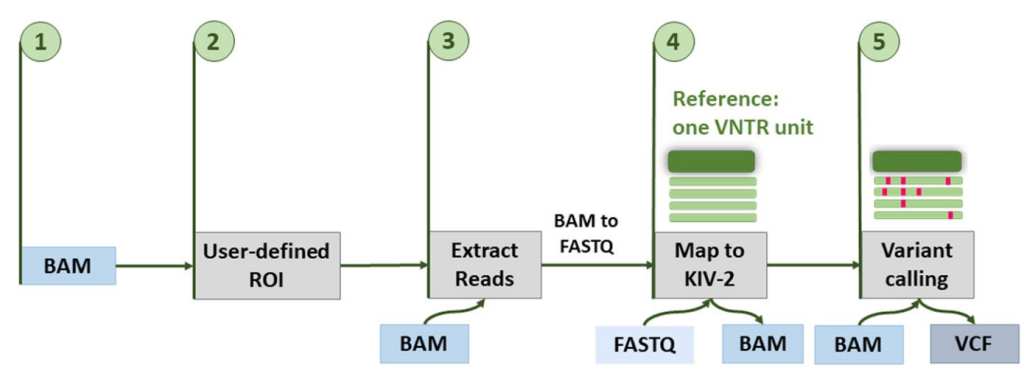
二、LPA KIV 2 VNTR变异检测流程构建及基准测试
对已知KIV-2变异(“金标准”)的24例样本进行WES测序,其中8例用于变异Calling管线调试(Dataset A),16例用于基准测试(Dataset B)。用于调试的8例样本分别进行WES v6kit(Dataset A experiment1)和v8kit(Dataset A experiment2)测序,16例基线测试样本只进行WES v8kit测序。
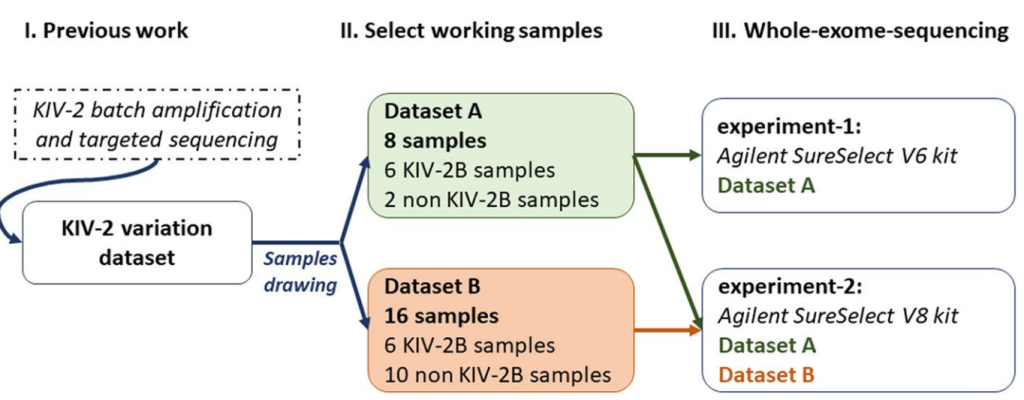
图8. LPA KIV 2 VNTR变异检测流程调试验证示意图
在金标准数据集中,数据集A中每个样本的编码KIV-2变异数量从4到24不等,数据集B中从2到17不等。大约80%的欧洲人包含KIV-2B单元,可以根据KIV-2 Exon 1的经典位置14、41和86的独特单倍型来识别(对应于参考序列中的位置594、621和666)。
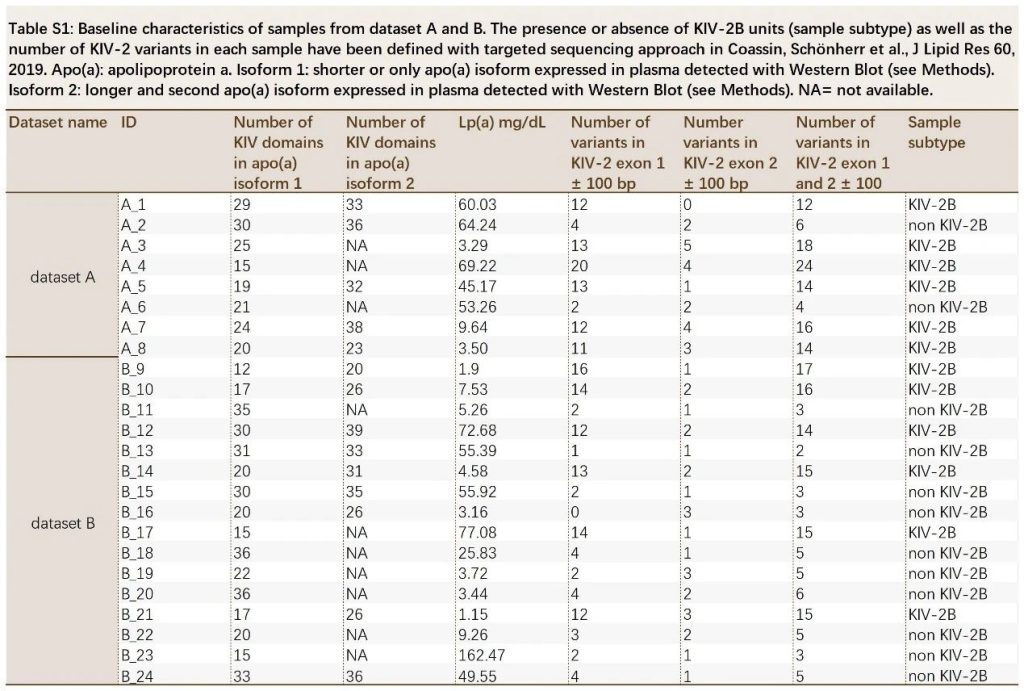
表2. 金标准数据集样本基本信息
按照KIV-2亚型不同(A、B和C),8例数据集A(调试)的2个样本和16例数据集B(基线测试)的10个样本被分类为非KIV-2B亚型样本。数据集B中较多的非KIV-2B样本允许研究在KIV-2 VNTR的变异检测工作流程中考虑KIV-2B亚型存在/缺失的策略。
首先,通过将所有可用的WES Reads比对到单个KIV-2重复单元来测试一个简单的KIV-2变异检测方法。然而,由于KIV-2的广泛同源性,许多来自其他KIV以及其他同源区域(例如PLG,LPAL2)的读取也非特异性的比对,并生成了来自PSVs的假阳性变异Calling。
在数据集A的实验1中,每个样本超过200个假阳性变异被Calling。这表明有必要将Reads提取限制在KIV-2 VNTR。因此,系统地评估了使用9个不同ROI(ROI-1到ROI-9)中KIV-2变异检测性能(图9)。
F1分数描述了precision(真阳性/(真阳性+假阳性))和sensitivity(真阳性/(真阳性+假阴性))的调和平均值,反映了假阴性和假阳性检测性能,分数越高,表示性能越好。
如表3所示,与ROI-1相比,使用ROI-2至ROI-7已经显著改善了变异Calling,数据集A的平均F1范围从31.0%到70.1%。当仅提取严格映射到KIV-2 VNTR(ROI-8)的读取而不包括侧翼区域时,实验1的平均F1值进一步提高到81.9%,实验2提高到84.4%。这表明提取区域对变异检测有重大影响,并且更严格的坐标限制在VNTR的起始和终止坐标,可以减少假阳性Calling并提高变异检测性能。
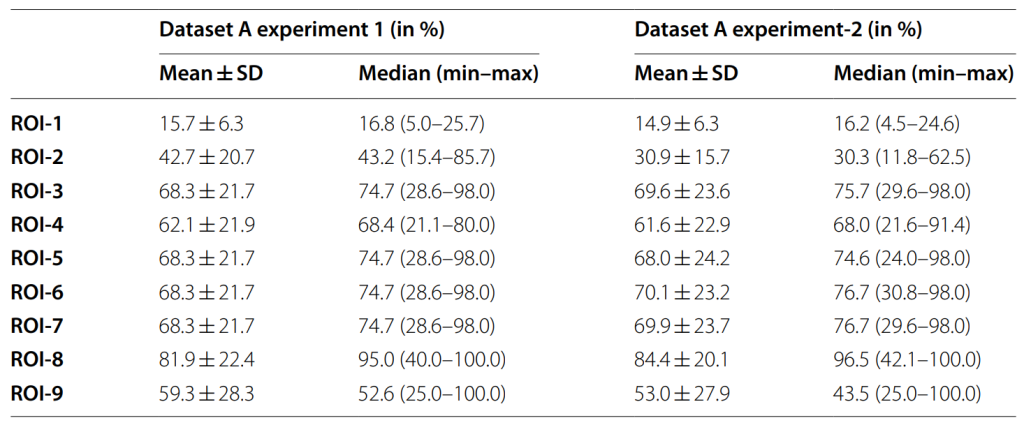
表3. 不同ROI的F1表现
然而,ROI-8的结果在两个实验中仍显示出个体水平上的较大差异,标准差(SD)为21.5–24%。因此,进一步调查了数据集中潜在的分层。
三、亚型特征改进了 LPA KIV-2 VNTR 的变异检测
在参考基因组hg19和hg38中,KIV-2 VNTR区域由6个KIV-2重复序列组成,其中包含5个KIV-2A单元和仅1个KIV-2B单元。因此,KIV-2B亚型错误地表现为固定且仅有一份拷贝,而实际上在人群中,个体内部的KIV-2B单元频率变化极大,甚至存在许多不携带KIV-2B重复单元的“非KIV-2B个体”。
我们注意到,ROI-8在KIV-2B个体中的表现最佳,两次实验的F1值分别达到94.2%和95.0%。然而,在非KIV-2B个体中,实验1的平均F1值降至45.0%,实验2降至52.3%,并导致大量假阳性变异(表4)。
与ROI-8相比,ROI-9不提取比对到KIV-2B亚型第一个外显子的Reads。因此,在非KIV-2B个体中,ROI-9在提取Reads时跳过参考基因组中的KIV-2B域,从而减少了假阳性Calling。ROI-9将非KIV-2B个体在两次测序实验中的F1值分别提高到95.5%和100%(表4)。
由于非KIV-2B个体中不存在真正的KIV-2B Reads,这些个体中比对到参考基因组KIV-2B亚型序列的Reads必定来自高度同源的区域,最可能来自与KIV-2B几乎相同的KIV-3。因此,进一步研究了在Nextflow分析流程中动态考虑KIV-2B亚型存在与否的策略,以最小化假阳性率。
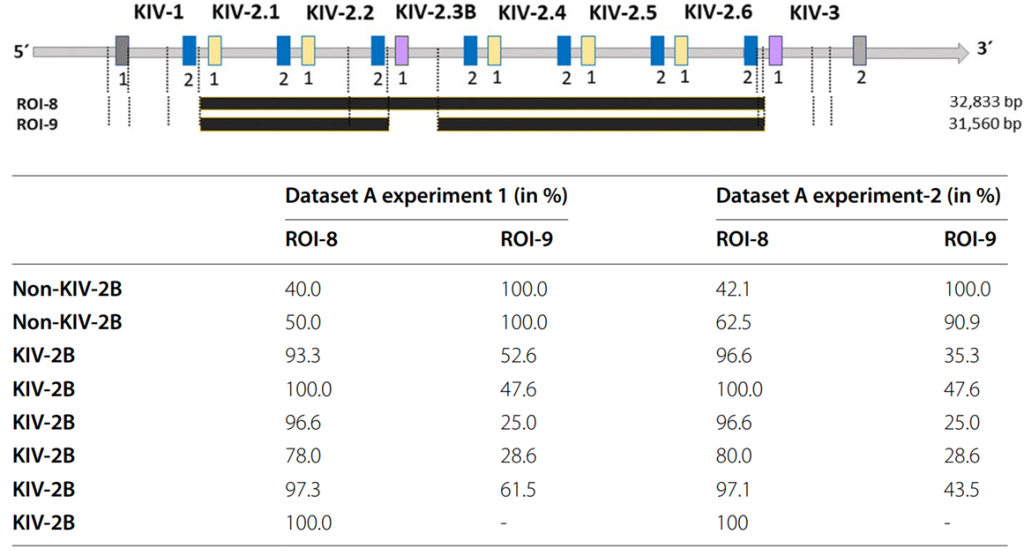
表4. KIV-2B亚型和非KIV-2B亚型个体中ROI-8和ROI-9变异检测性能差异
上述实验表明,ROI-8和ROI-9分别是KIV-2B和非KIV-2B样本变异检测的最佳策略。因此,需要寻找一种独特的序列特征,以便在Reads提取之前无差别地区分KIV-2B和非KIV-2B个体,并动态分配最佳ROI。
尽管文献中报道KIV-3的第一个外显子与KIV-2B的第一个外显子相同,但通过一代测序发现,在KIV-3第一个外显子的86号位置发现了一个SNP(称为“特征位置”),可以区分KIV-2B和非KIV-2B个体。
如图6A所示,所有KIV-2B样本在特征位置至少携带一个T-等位基因(这也是基因组中的参考碱基),而几乎70%的非KIV-2B个体(16个样本中的11个)在该位置携带C/C纯合基因型。这表明KIV-2B重复单元的存在与KIV-3外显子1的86号位置(T-特征)的T-等位基因之间,以及KIV-2B单元的缺失与该位置的C-等位基因之间存在相关性。
因此,定义了一个KIV-2B特征序列(CCACTGTCACTGGAA),该序列包含KIV-3外显子1的86号位置的T-等位基因。假设测序读取中此特征序列的存在或缺失可以区分样本亚型(KIV-2B和非KIV-2B),并将每个样本分配到最佳ROI进行读取提取。
使用非KIV-2B样本的现有WES数据(n=12)来计算特征序列出现次数的经验阈值,以区分这两种样本亚型,并动态分配每个样本到ROI-8和ROI-9中进行分析,该方法大规模提高了KIV-2 VNTR区域的变异检测准确性。
在两次实验中,动态ROI分配的平均F1分数均约为95%。数据集A包含两个非KIV-2B样本,使用动态ROI分配后,实验1的平均F1分数提高了16.8%(从81.9%增至95.65%),实验2提高了12.7%(从84.4%增至95.14%),同时样本间的变异程度也降低了。重要的是,基于特征序列的方法在非KIV-2B样本中未产生假阳性变异,而使用静态ROI-8方法最多可产生12个假阳性变异。
将动态特征方法应用于验证数据集B(n=16)时,F1 从 63.2%提高到 90.8%(+43.6%;仅与 ROl-8 相比),而且样本之间的差异较小。
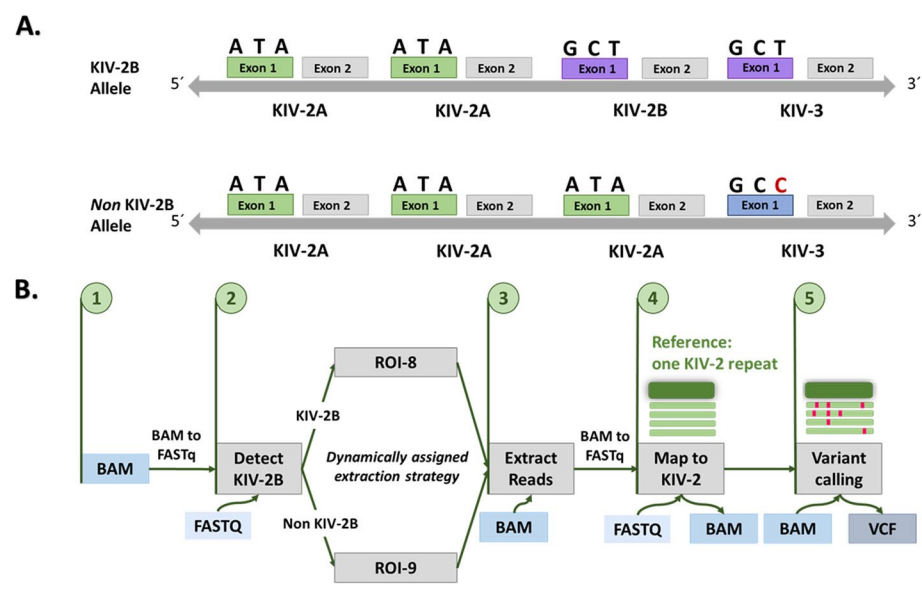
图10. 利用KlV-2B 单倍型与 KlV-3 第一个外显子86位基因型之间的连锁不平衡关系动态分析不同个体KIV-2变异
四、在UK Biobank中检测到 LPA KIV-2 变异情况
通过利用KIV-2特征的动态个体化分析流程流程,分析了UK Biobank全外显子测序(UKB 200K WES发布)中的199,119个样本中的KIV-2 VNTR。
在75%的样本中(n=149,184)检测到KIV-2B特征序列,具有黑人血统的个体显示出最低比例的KIV-2B个体,这与先前的发现一致。在所有样本中检测到了总共707个独特的突变,其中包括256个错义突变、37个无义突变和8个剪接位点突变。有95个内含子突变位于外显子周围25bp的区域内(称为“剪接区域”),可能影响内含子的核心剪接元件。
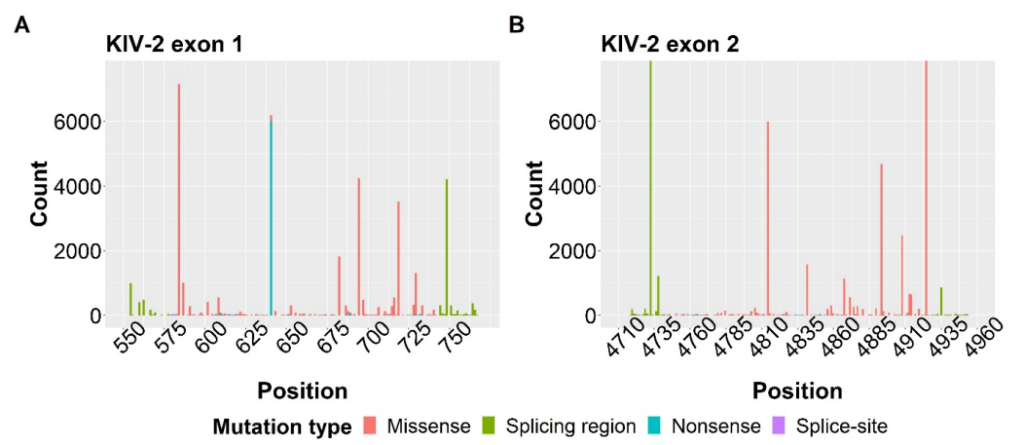
图11. KIV-2变异检出及个体数量
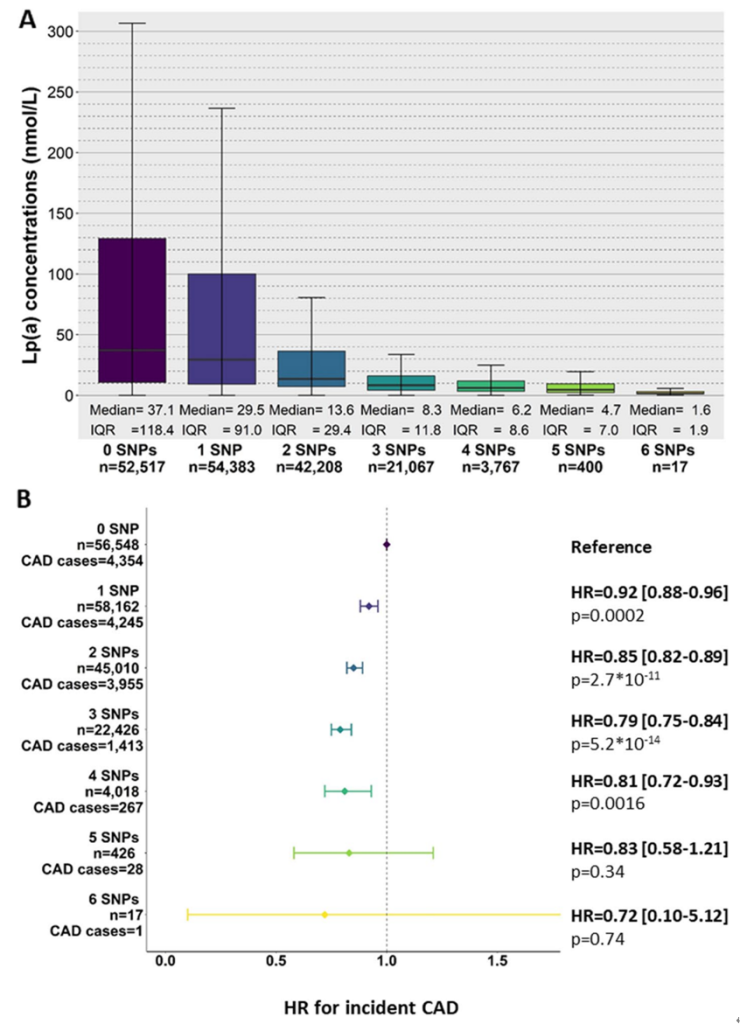
在具有白人血统的UKB参与者中鉴定了9个与Lp(a)浓度降低显著相关的SNP(p值<5×10−8),携带者频率>0.015。这些SNP包括先前鉴定的R21X(位置640)、4733G>A和4925G>A,以及KIV-2 VNTR中的6个其他SNP(在此参考序列中的位置515A>G、516A>G、584C>T、4672A>G、4698C>A、5013G>T)。
随着效应等位基因数量的增加,Lp(a)浓度中位数从参考组(仅参考等位基因,n=52,517)的37.1 nmol/L(IQR=118.4 nmol/L)下降到携带5个降低Lp(a)等位基因的个体(n=400)的4.7 nmol/L(IQR=8.6 nmol/L),甚至在17名携带6个降低Lp(a)等位基因的个体中达到1.6 nmol/L(IQR=1.9 nmol/L)(图12A)。
这些SNP还显示出对冠心病(CAD)额外的保护作用。与非携带者(n=56,548,CAD病例=4354)相比,随着降低Lp(a)等位基因数量的增加,CAD事件发生风险逐渐降低。四个降低Lp(a)的SNP(n=3767,CAD病例=1413)将CAD风险降低了约19%(HR=0.81,95%置信区间(CI)=0.72–0.93],p值=0.0016)(图12B)。
讨论
NGS技术的发展为基因组研究带来了巨大的飞跃,尽管第三代测序技术的长读长已经出现,NGS仍然是大多数人口规模测序项目的主力。研究人员开发了一种从短读长测序数据中检测VNTR中变异的方法,并提供了一个可扩展和并行化的Nextflow管道。
为了优化变异检测方法,使用了LPA基因中的大型KIV-2 VNTR,由于其与邻近KIV以及其他同源基因的广泛同源性,这使得其特别具有挑战性,因此是设置和基准测试的理想候选者。
在英国生物样本库(UKB)中测试了这一方法,并发现了一组近300个高置信度的影响蛋白质序列的变异(包括37个无义突变和8个剪接位点突变),其中包括多个先前的发现,这进一步支持了该变异识别的有效性。此外,这也揭示了隐藏在KIV-2中的SNP与降低的Lp(a)浓度之间的新关联,并将迄今为止最大的KIV-2变异数据集作为返回结果提供给UKB。
在广泛使用的变异检测格式(VCF)中表示VNTR仍然是一个持续的挑战。未来的工作需要开发一种新的文件格式或增强现有的VCF文件格式,以便准确表示VNTR,以及创建新的VNTR基准测试工具。
参考资料
1. Tsimikas S, Bittner V. Particle Number and Characteristics of Lipoprotein(a), LDL, and apoB: Perspectives on Contributions to ASCVD. J Am Coll Cardiol. 2024 Jan 23;83(3):396-400.
2. Coassin S, Kronenberg F. Lipoprotein(a) beyond the kringle IV repeat polymorphism: The complexity of genetic variation in the LPA gene. Atherosclerosis. 2022;349:17-35.
3. Mukamel RE, Handsaker RE, Sherman MA, et al. Protein-coding repeat polymorphisms strongly shape diverse human phenotypes. Science. 2021;373(6562):1499-1505.
4. Di Maio, S., Zöscher, P., Weissensteiner, H. et al. Resolving intra-repeat variation in medically relevant VNTRs from short-read sequencing data using the cardiovascular risk gene LPA as a model. Genome Biol 25, 167 (2024).