


藏在“暗处”的基因密码:LDLR非编码区变异如何导致家族性高胆固醇血症?
- boke
- 2026-03-13
- 11:07 上午
家族性高胆固醇血症是一种遗传性疾病,患者从出生起就面临极高的“坏胆固醇”(LDL-C)水平,早发冠心病风险显著增加。长期以来,医学界将目光聚焦于LDLR、APOB、PCSK9这三个基因的编码区——那些直接决定蛋白质氨基酸序列的区域。
然而,约有一半临床诊断为FH的患者,在这三个基因的编码区中找不到任何致病变异。他们的问题究竟出在哪里?
2025年发表于《European Journal of Human Genetics》的一项研究,首次通过大规模全基因组测序和功能实验,揭示了一种全新的致病机制:LDLR基因5‘ UTR的变异,通过引入“冒牌”起始密码子,从根本上破坏了LDL受体的正常合成。
一、什么是5‘ UTR?一个被忽视的调控区域
每个基因的编码区前都有一段被称为“5’非翻译区”(5’ UTR)的序列。它不编码蛋白质,却像一个精密的“翻译调控开关”——核糖体需要先扫描这一段区域,找到正确的起始密码子(AUG)才能开始蛋白质合成。
正常的5‘UTR中通常不会出现AUG密码子。但如果发生变异,在5’UTR中意外地产生了一个新的AUG(称为“上游起始密码子”),就会发生以下情况:
核糖体在扫描时优先识别了这个“冒牌“的起始密码子,开始翻译一段无意义的短肽,而真正的LDLR编码序列被忽略。结果就是,正常的LDL受体蛋白生产大幅减少。
二、研究发现了什么?
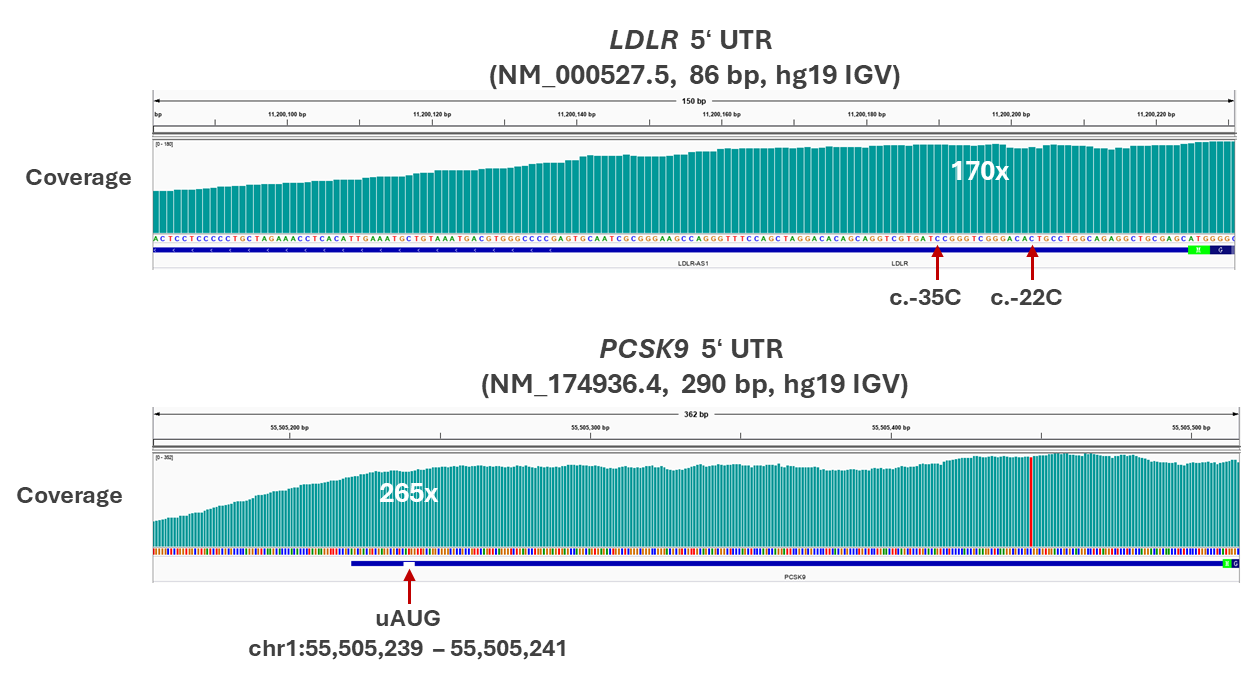
研究团队对英国10万人基因组计划中的536名FH患者进行了全基因组测序,并运用专门的生物信息学工具UTRannotator对5‘UTR区域的变异进行筛选[1]。他们重点关注能够创建或破坏上游起始密码子的变异。
1
两个关键变异的发现

c.-35C>G:一个全新的发现
在一名FH患者中,研究团队发现了一个位于LDLR基因5‘ UTR的变异:c.-35C>G。该变异在正常人群数据库中极为罕见(798,530人中仅1例携带),且预测会在5’ UTR中创建一个与正常阅读框不一致的上游开放阅读框,长度为63个氨基酸。
c.-22del:一个被重新认识的变异
研究团队还对2005年文献中报道过的c.-22del变异进行了重新评估[2]。该变异同样预测会创建一个新的上游起始密码子和58个氨基酸的上游开放阅读框。该变异最初在一个土耳其FH家系中被发现:先证者为复合杂合子,年仅8岁就出现黄色瘤和冠心病,总胆固醇高达20.12 mmol/L;其父亲携带c.-22del变异,表现为杂合子FH,总胆固醇8.83 mmol/L。
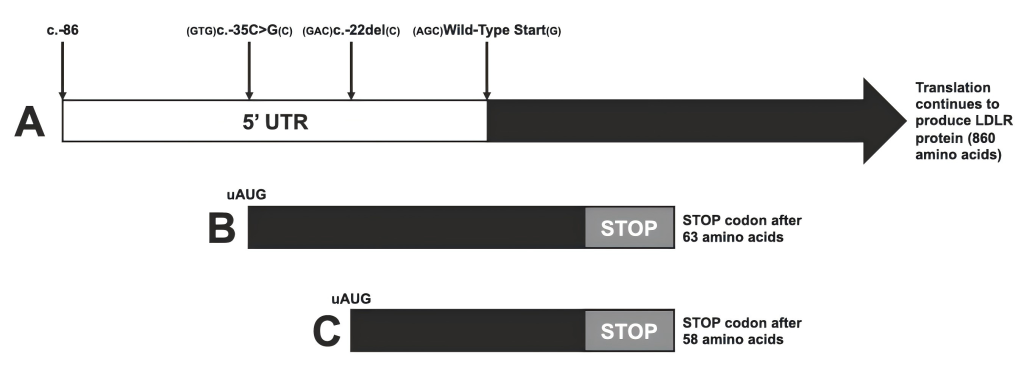
三、功能实验:证实致病机制
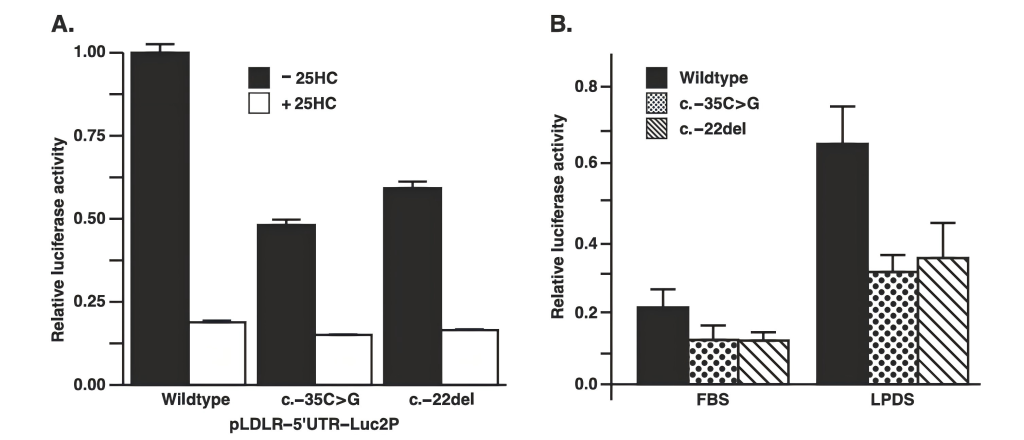
为验证这两个变异是否真的影响LDLR表达,研究团队设计了精密的荧光素酶报告基因实验。他们将野生型和突变型LDLR 5‘UTR序列分别连接在报告基因前,转染细胞后检测报告基因的表达水平。
c.-35C>G变异使报告基因活性降低 59.4% ± 4.5%
c.-22del变异使报告基因活性降低 46.7% ± 3.4%
这意味着,这两个变异均导致LDLR蛋白表达显著下降,足以解释患者的FH表型。
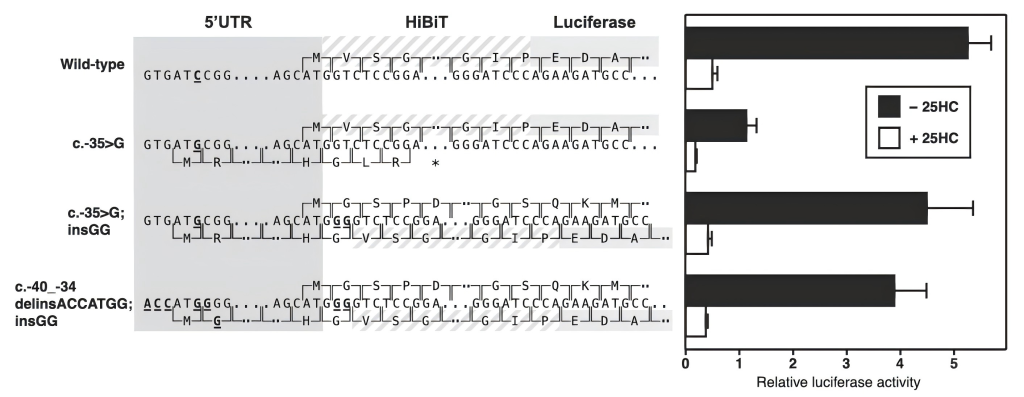
研究团队通过实验揭示了c.-35C>G变异的致病机制:该变异在LDLR基因5‘UTR中引入了一个与正常LDLR编码序列不同框的上游起始密码子(uAUG),导致核糖体优先从该uAUG开始翻译一段无义短肽,从而无法正常起始LDLR蛋白合成。实验证明,当通过人工构建使该uAUG与下游报告基因同框时(插入2个G),翻译效率反而显著增强,说明uAUG本身具备功能性翻译起始能力;进一步使用理想Kozak序列和同框-“c.-40_34 delinsACCATGG; insGG”也并未明显增强表达,进一步说明该变异的致病性关键在于阅读框不一致,而非Kozak序列的强弱。这一机制很好解释了c.-35C>G为何导致LDLR表达降低59.4%。即使没有理想的Kozak序列(翻译起始的增强序列),仅仅是上游起始密码子的存在就足以显著干扰正常翻译。
四、研究团队根据ClinGen指南对两个变异进行了系统性评估
按照ClinGen家族性高胆固醇血症变异解读专家小组的LDLR变异分类指南,研究团队对c.-35C>G和c.-22del两个变异进行了证据评估。荧光素酶功能实验显示,c.-35C>G变异使表达降低59.4% ± 4.5%,达到PS3_Supporting证据强度;而c.-22del变异降低46.7% ± 3.4%,未达到该标准。结合其他证据,c.-35C>G被归类为可能致病性,c.-22del仍为意义不明确 [3]。
值得注意的是,这两个变异的效应强度(46-59%表达降低)远超既往报道的LDLR启动子区变异(SP1结合位点变异仅降低6-26%,SREBP结合位点变异降低11-40%)。这说明,5’ UTR上游起始密码子获得型变异是FH的一种强效致病机制。
五、对临床实践的启示
1
扩大遗传检测范围
本研究强调,对于临床诊断为FH但编码区未检出致病变异的患者,应将检测范围扩展至5‘ UTR区域。传统的基因检测往往局限于外显子和邻近剪接区,可能遗漏这类位于非编码区的致病性变异。研究团队明确指出:“我们的发现强调了在FH遗传诊断中考虑非编码区变异的重要性。”
2
重视上游开放阅读框的影响及基因诊断的价值
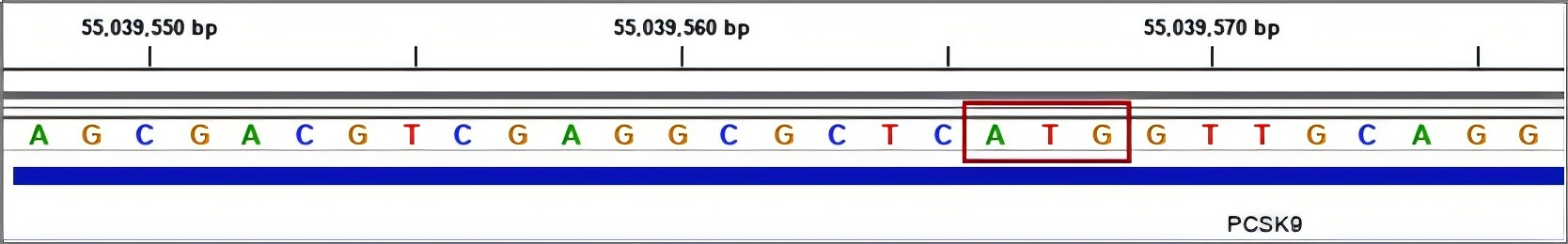
上游开放阅读框的引入是一种值得关注的新型致病机制。研究团队指出,PCSK9基因本身也编码一个上游开放阅读框,其上游AUG位于Chr1: 55039566-55039568,hg38 (chr1:55,505,239 – 55,505,241, hg19)。“uORF功能丧失型变异被认为与翻译增强相关”,即PCSK9的GOF变异,这种变化可能导致LDL受体降解增加,LDL-C水平升高。
鉴定出可能致病的uAUG变异可以为患者提供基因诊断,进而用于家系级联筛查,并根据患者的血脂水平制定个体化的降脂治疗方案(如他汀类药物剂量优化、联合用药选择等)。虽然目前尚无针对uAUG变异本身的特异性疗法,但明确LDLR表达降低的机制,为未来可能的RNA靶向治疗或基因治疗提供了理论基础和研究方向。
六、研究的局限性
1. 实验结果未与536名FH患者的血脂水平进行直接关联分析。
2. c.-35C>G变异的携带者已失访,无法获取家系样本进行进一步的共分离研究。
3. 未通过Western blot直接检测LDLR蛋白表达水平,进行蛋白水平验证。
尽管如此,功能实验的结果已经为这两个变异的致病性提供了有力证据。
七、未来展望
本研究开启了一个新的研究方向:系统筛查FH患者的非编码区变异。随着全基因组测序成本的下降和生物信息学工具的完善,未来有望发现更多类似的上游起始密码子获得型变异。
研究团队呼吁:
“未来的研究应聚焦于扩展FH患者5‘ UTR变异的目录,并利用体外和体内模型系统研究其功能影响。这不仅有助于解决目前约半数FH患者‘基因阴性’的困境,也能更深入地理解LDLR基因的调控机制。”
基因的“暗物质”正在被照亮
人类基因组中,编码蛋白质的区域仅占约2%,其余98%为非编码区。长期以来,这些区域被视为基因组的“暗物质”,其功能和作用机制充满未知。
这项研究揭示了一种存在于“暗处”的致病机制——5‘ UTR变异通过引入上游起始密码子,从根本上破坏LDLR蛋白的正常生产。它不仅为约半数“基因阴性”的FH患者提供了新的诊断方向,也提醒我们:在遗传病的诊断中,关注那些不起眼的非编码区,或许能发现意想不到的答案。
对于FH患者及其家庭而言,这意味着更多未解之谜有望在未来被解开,更多家庭将有机会获得明确的基因诊断,从而实现早期干预和精准治疗。
伯科单基因遗传性心血管病Gene Panel模块覆盖148+基因,涵盖了《单基因遗传性心血管疾病基因诊断指南》(2019年)、《常见单基因遗传性心血管病基因变异致病性分析中国专家共识》(2024年)、《遗传性心血管疾病基因检测和遗传咨询中国专家共识》(2024年)等多部相关指南/共识推荐基因,所有基因均覆盖了UTR区域,在实际应用中对UTR区域覆盖优异,满足对非编码区的临床科研需求。
参考文献:
1.Bird M, Tung CJ-P, Pittman AM, et al. Novel start codon variant in the 5‘ UTR of LDLR associated with familial hypercholesterolaemia. Eur J Hum Genet. 2025. doi:10.1038/s41431-025-01893-y
2.Szolen MM, Whittall R, Oner C, et al. The molecular basis of familial hypercholesterolaemia in Turkish patients. Atherosclerosis. 2005;180:63-71.
3.Szolen MM, Whittall R, Oner C, et al. The molecular basis of familial hypercholesterolaemia in Turkish patients. Atherosclerosis. 2005;180:63-71.