


Cell | 遗传修饰因子、验证偏倚与复杂疾病的表型差异
- boke
- 2025-10-16
- 10:21 上午
为什么两个携带完全相同 “致病基因变异” 的人,一个可能出现严重智力障碍,另一个却能正常生活?这一 “变异外显度差异” 的谜题,正是遗传学领域的核心挑战。
2025 年发表在《Cell》的一项研究,通过分析 2455 名携带致病性主变异体的个体(涵盖 16p12.1 缺失、16p11.2 CNV、CHD8 等 SNV),结合 8 个不同验证方式的队列,首次系统性揭示:
修饰变异体(罕见 SNV/CNV/STR 扩增)与多基因风险评分(PRS,反映全基因组常见变异累积效应)通过 “累加 / 协同作用” 共同修饰主变异体表型,效应具有主变异特异性、表型特异性、验证方式特异性;
16p12.1 缺失的基因网络 “分散性”(核心基因与基因组其他基因的 “交互连接模式” 松散且无集中方向)是其表型异质性高于 16p11.2 缺失的关键机制,而PRS对这一异质性的调控发挥了重要作用;
验证偏倚导致健康队列(如 UKB)与疾病队列(如 DD)的修饰/修饰变异体负荷反向,PRS同时参与解释不同队列的表型差异;
主 – 修饰变异交互作用是表型异质性的核心驱动因素。
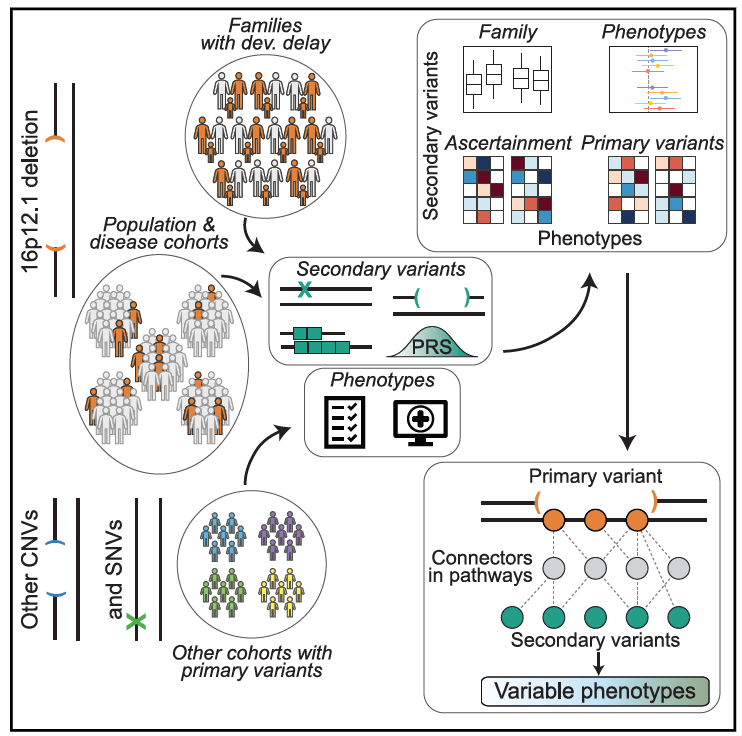
一
研究背景:从 “单基因致病” 到 “多因素协同”,表型异质性的临床与科学挑战
临床现象:表型异质性普遍存在
相同致病变异(如 16p12.1 缺失)的携带者表型差异显著——93% 的 16p12.1 缺失患病儿童从表型轻微的父母那里传该变异,表型覆盖 “严重自闭症 + 智力障碍” 至 “完全健康”。
传统认知局限
既往认为 “遗传病=单个高外显率基因变异”,但实际中,传统 “单基因致病” 模型无法解释孟德尔疾病的外显率(患病概率)与外显度(症状严重程度)差异,仅少数研究提出 “多重遗传诊断” 或 “多基因风险修饰”,但未系统验证PRS 在其中的协同作用。
既往理论基础:“二次打击模型”,疾病发生的核心逻辑
——第一次打击(主变异)
如 16p12.1 缺失,仅使个体 “易感疾病”,不直接导致严重症状,相当于 “在基因组堤坝上凿开缺口”,提高风险但不必然 “溃堤”。
——第二次打击(修饰/修饰变异体)
基因组中其他罕见变异(如错义突变、STR 扩增)、常见变异的累积效应(多基因风险评分 PRS),前者通过干扰局部通路细化表型特征,后者通过全基因组微效位点的累加,影响疾病发病阈值与症状严重程度;相当于 “在缺口处叠加冲击”,最终决定症状严重程度。
——典型案例
同时携带 MKS1(梅克尔 – 格鲁伯综合征相关)和 BBS1(巴德特 – 比德尔综合征相关)变异的个体,会协同导致两种疾病均不典型的癫痫症状,而该症状的严重程度,进一步与精神疾病相关 PRS 存在关联。
核心科学问题:为何携带相同主变异体的个体表型差异显著?修饰变异体的“类型/功能”以及队列“验证方式”偏好的影响如何?
研究目的:通过多队列、全基因组技术,系统解析修饰变异体与验证偏倚对复杂疾病表型异质性的驱动作用,填补 “主 – 修饰变异交互机制” 的研究空白。
二
研究设计:多队列 + 全基因组技术,全面捕捉遗传修饰效应
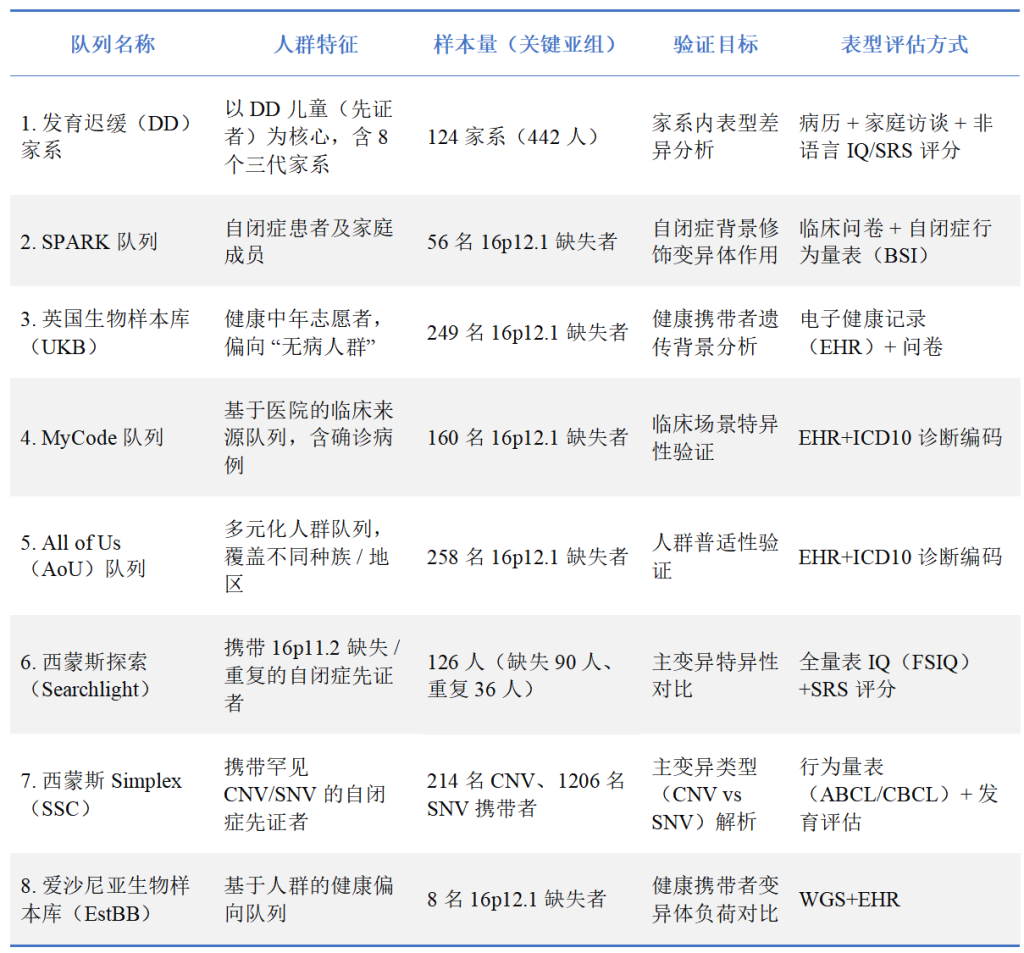
1. 队列设计:覆盖 “疾病 – 健康” 全光谱,对比验证偏倚
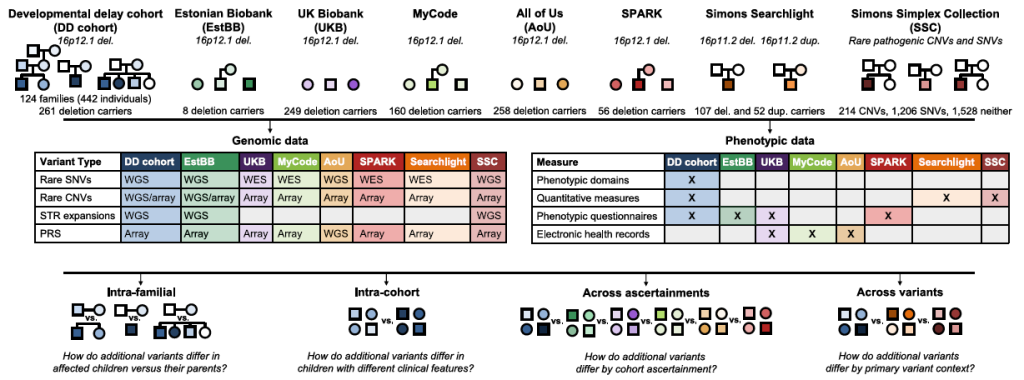
Figure 1. Secondary-variant-phenotype analyses in 2,455 individuals with primary pathogenic variants.
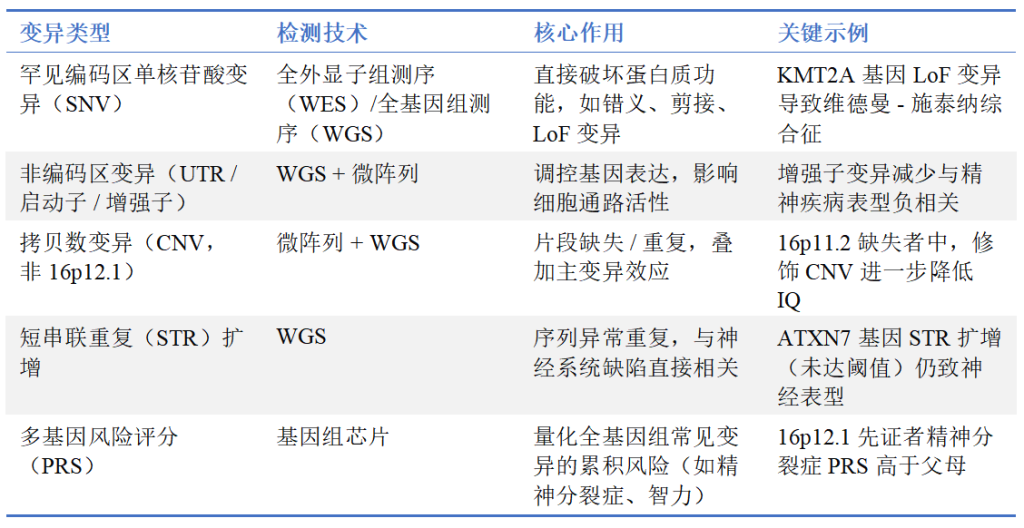
2. 技术方法:全基因组级 变异检测“捕捉所有变异”
三
核心结果:从 “家系” 到 “多队列”,层层解析机制
1
1. 第一步:家系分析 —— 先证者症状重,与修饰变异体负荷及 PRS 特征相关
1)16p12.1 缺失携带者表型异质性量化
先证者 vs 携带缺失的父母:非语言 IQ(Intelligence Quotient,智商) 降低 1.98 个标准差(p=2.13×10⁻⁵),社交障碍(SRS 评分)升高 1.91 个标准差(p=2.59×10⁻⁷),自闭症特征显著;同时,先证者精神分裂症 PRS 高于父母(p=0.009),头围减小(p=0.001)、BMI(Body Mass Index,身体质量指数) 升高(p=0.009);
先证者 vs 其他队列先证者:16p12.1 缺失先证者 IQ 比 SSC 自闭症先证者低 1.06 个标准差(p=0.004),SRS 评分比 16p11.2 CNV 先证者高 0.96 个标准差(p=8.31×10⁻⁶);
发育里程碑延迟:先证者翻身、说话、走路等里程碑均显著延迟(p<0.05),与 DD 队列验证目标一致。
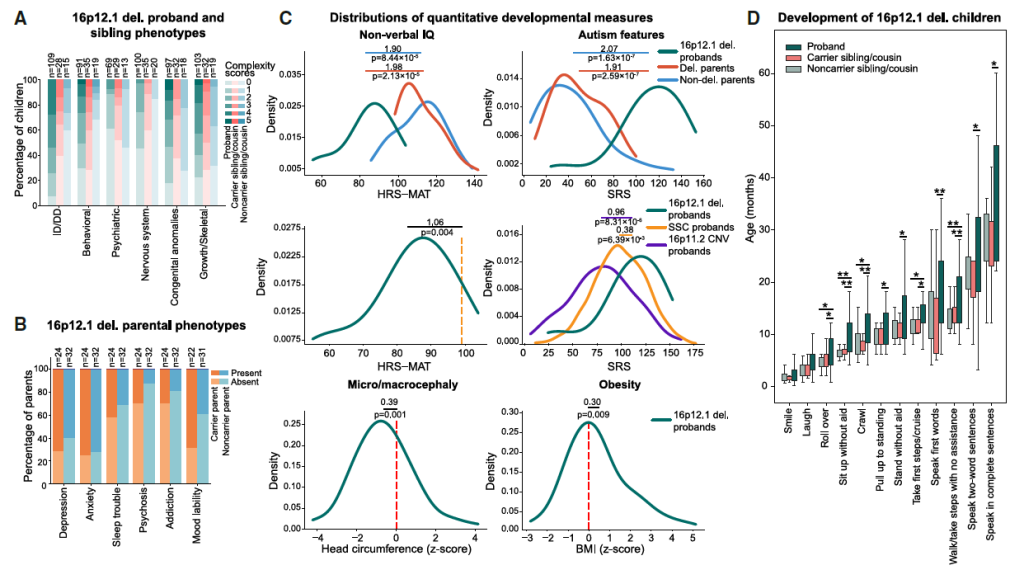
Figure 2. Variably expressive phenotypes of family members with the 16p12.1 del.
2)修饰变异体与PRS的累积证据
先证者比携带缺失的父母多携带 23% 的错义(LF)变异(p=0.034,LF 指受进化约束的有害变异);
三代家系中观察到 “变异负荷逐代增加 + 表型逐代加重” ,同时伴随 PRS 的逐代升高(如 GL_077 家系:祖母轻度认知异常→母亲抑郁焦虑→先证者严重神经发育障碍,精神分裂症 PRS 同步上升);
先证者与不携带缺失的父母相比,功能缺失型(LF)变异体数量增加(p=0.028);31% 的先证者(31/99)至少携带 1 种额外致病变异(如 ClinVar 致病性变异、DBD 数据库 LoF 变异),其中 7 名先证者携带明确致病性变异。
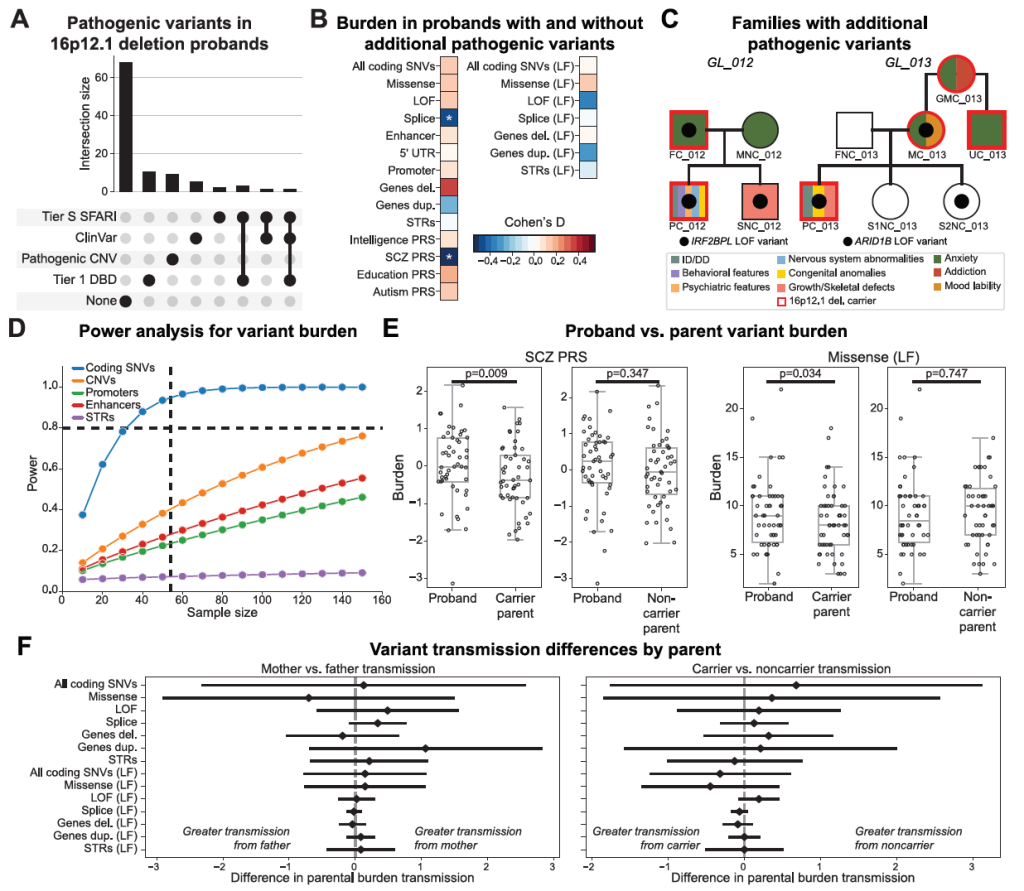
Figure S2. Disease association, statistical power, and burden of secondary variants, related to Figure 3
3)变异功能特异性富集
修饰变异体集中在 “大脑发育关键基因”,包括:
胎儿期腹外侧额叶皮质(p=3.32×10⁻⁸)、海马体(p=2.33×10⁻⁷)高表达基因;
成人运动皮质的兴奋性(p=1.22×10⁻⁴)和抑制性神经元(p=2.04×10⁻²⁴)高表达基因;
与 16p12.1 基因(如 VWA3A、CDR2)在神经祖细胞(NPC)中共表达的基因(p=0.017~1.27×10⁻²²),证明其精准干扰主变异相关通路;
同时,PRS 关联基因多涉及全脑神经元信号通路(如突触传递、神经元分化),与修饰变异体的局部通路作用形成互补。
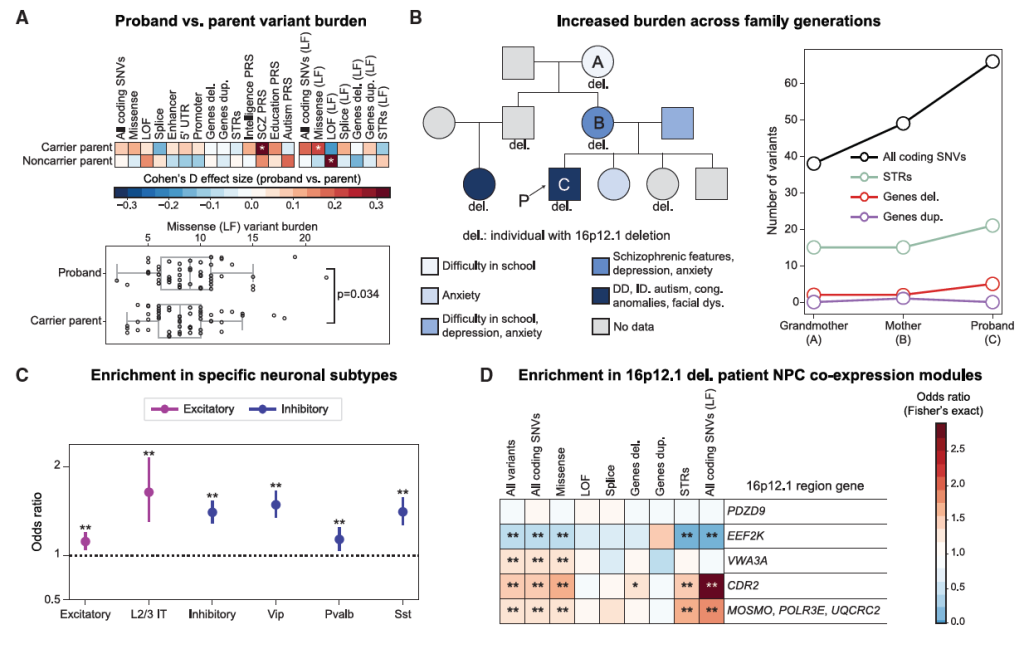
Figure 3. Secondary variants contribute to phenotypic variability within 16p12.1 del. Families
2
第二步:机制解析 ——16p12.1 缺失 “表型多变” 的核心原因:基因网络 “各自为战”
1)16p12.1 与 16p11.2 基因网络差异:
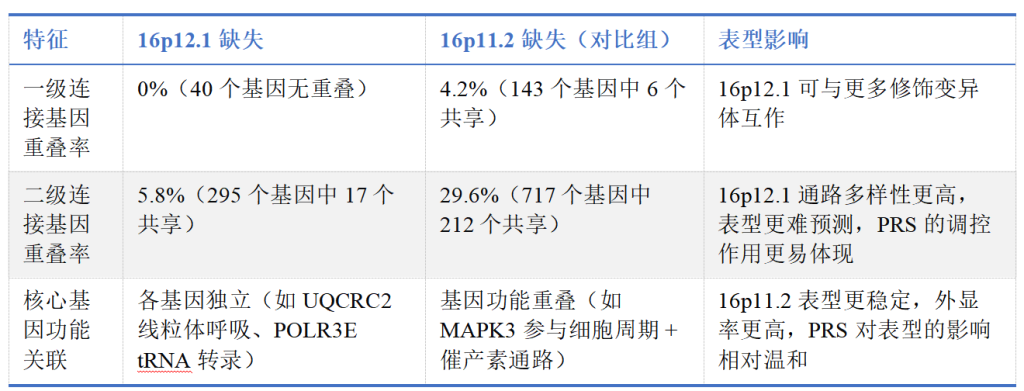
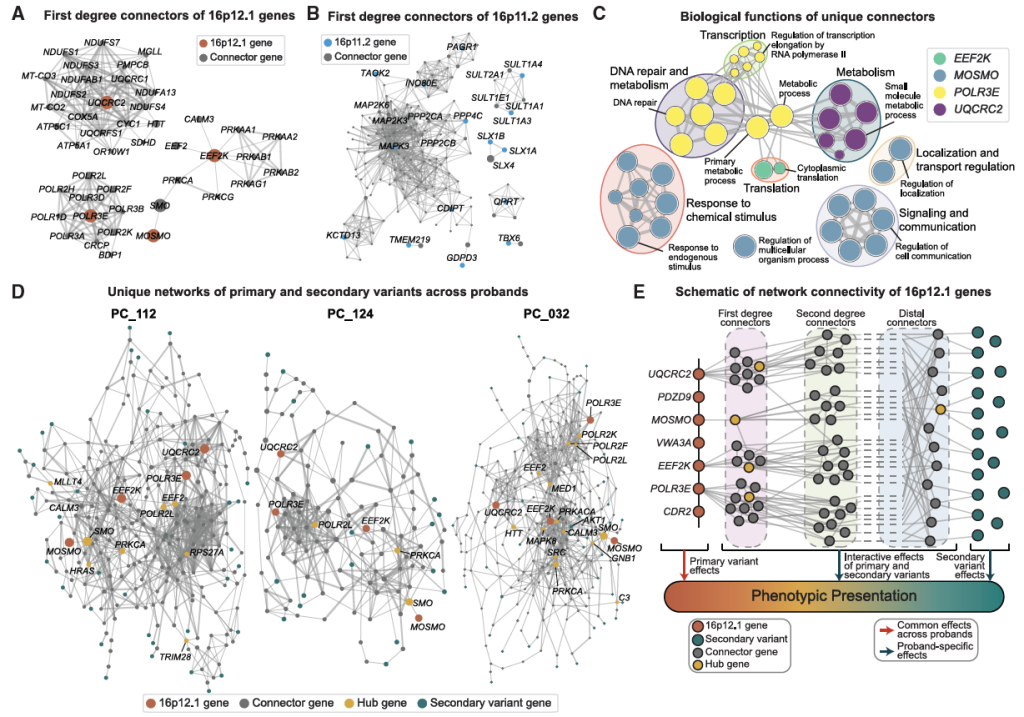
Figure 4. Connectivity of primary and secondary variants in a gene interaction network
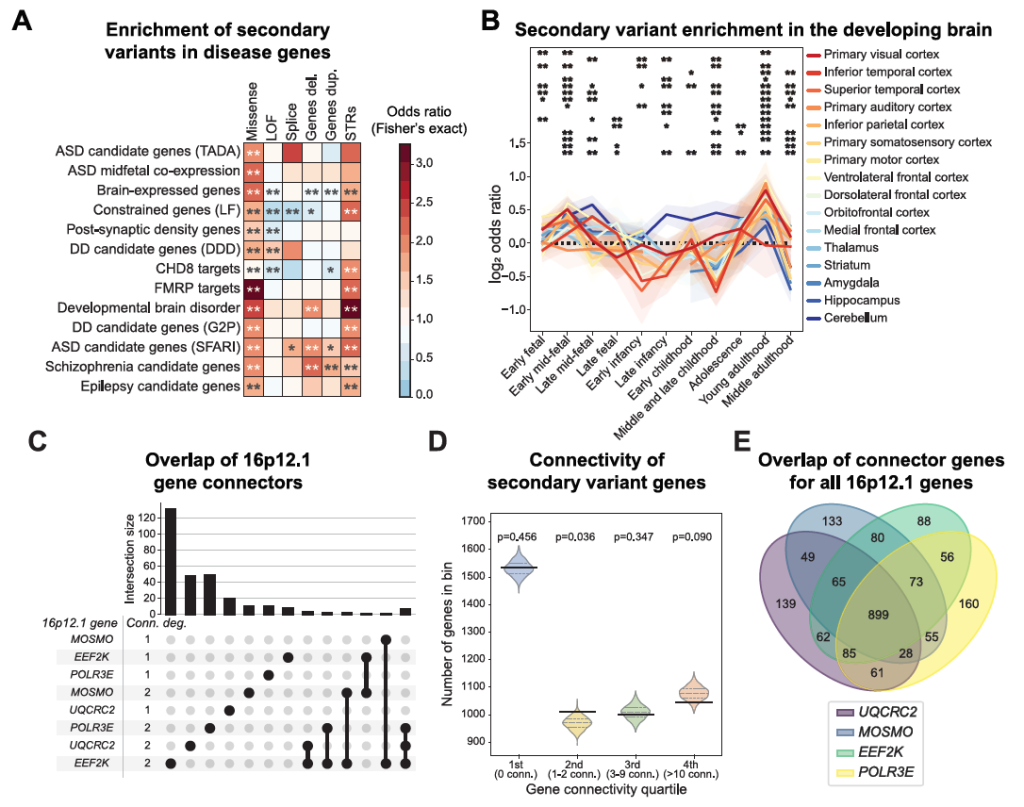
Figure S4. Functional effects and network connectivity of secondary variants observed in 16p12.1 del. probands, related to Figures 3 and 4
2)先证者特异性核心基因
即使表型相似(如均患自闭症 + 智力障碍),不同先证者的 “基因互作核心” 不同:
仅患自闭症的先证者(PC_112):核心基因为 Ras 通路的 HRAS(与自闭症相关),其表达水平与自闭症 PRS 存在关联;
仅患智力障碍的先证者(PC_032):核心基因为 PI3K-AKT-mTOR 通路的 AKT1(神经发育关键),智力 PRS 较低可能进一步加剧 IQ 降低;
证明 “个体特异性修饰变异体+PRS特征” 共同导致表型异质性。
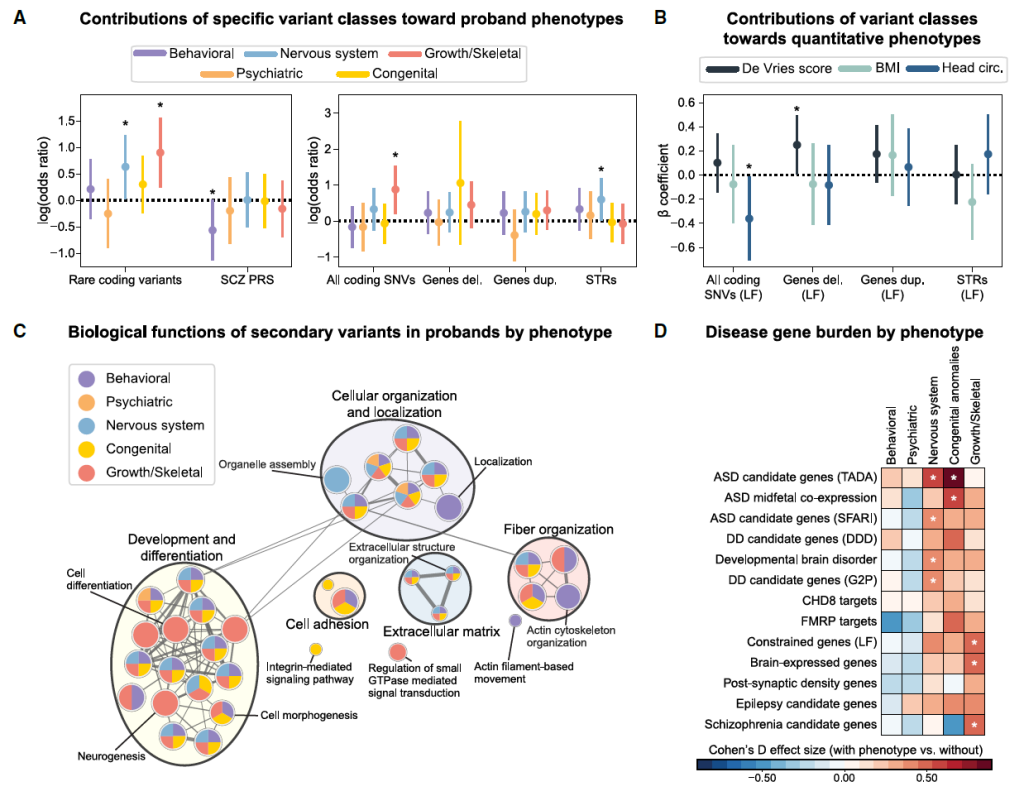
Figure 5. Secondary variant associations for phenotype domains of 16p12.1 del. probands
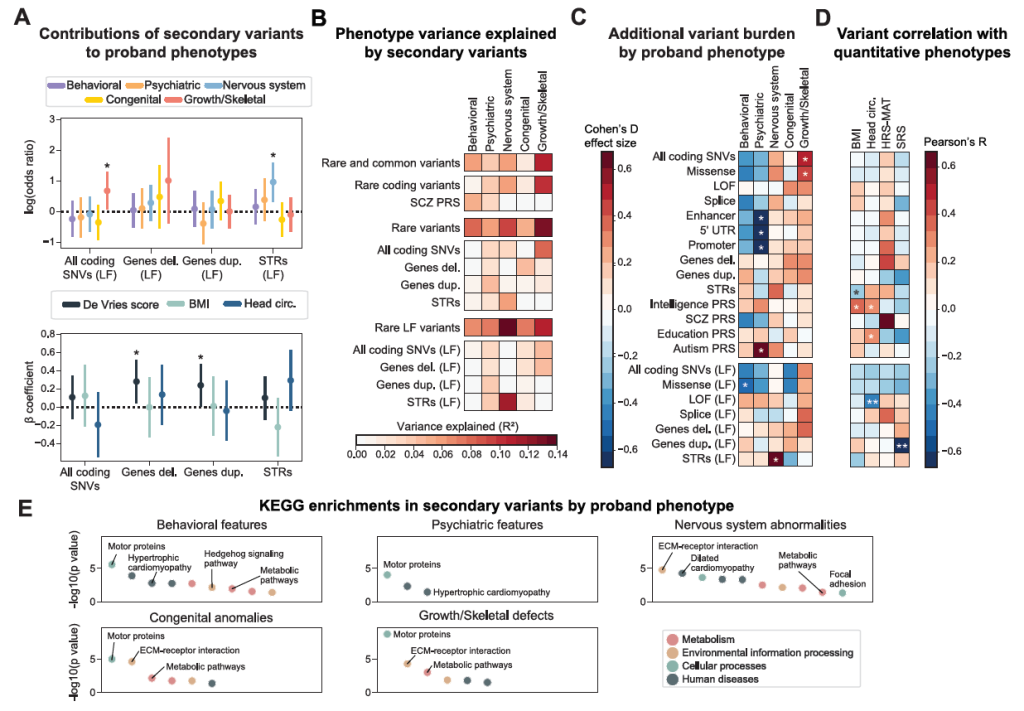
Figure S5. Secondary variant associations with 16p12.1 del. phenotypic domains, related to Figure 5
3
第三步:多队列对比 —— 验证偏倚 “反转” 结果:健康与疾病队列的 “遗传背景差异”
1)表型差异:队列来源决定 “症状谱”
UKB(健康偏向):16p12.1 缺失携带者主要表型为代谢疾病(肥胖:BMI 升高 1.23,p=0.006)、高血压,神经症状罕见,代谢相关 PRS 相对较高(p=0.003);
DD/AoU(疾病偏向):携带者主要表型为神经发育障碍(智力迟缓、自闭症),焦虑患病率是 UKB 的 3~5 倍(p=9.80×10⁻⁵~2.73×10⁻⁵),神经疾病相关 PRS 显著高于 UKB 队列。
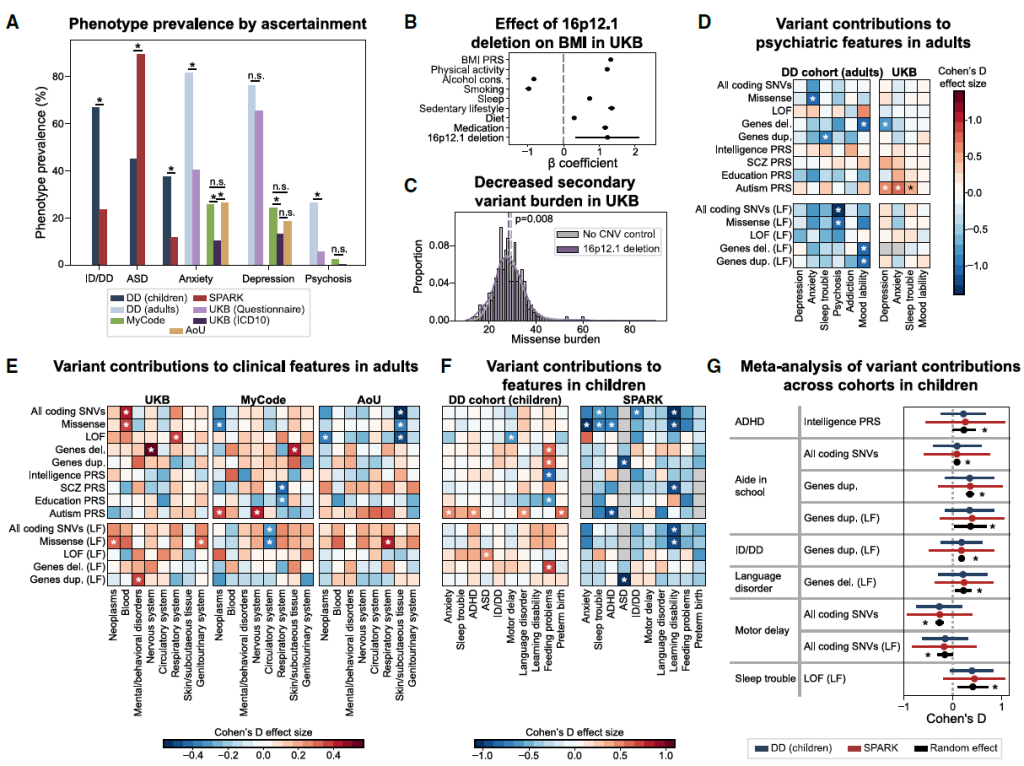
Figure 6. Effects of ascertainment on genotype-phenotype associations of 16p12.1 del.
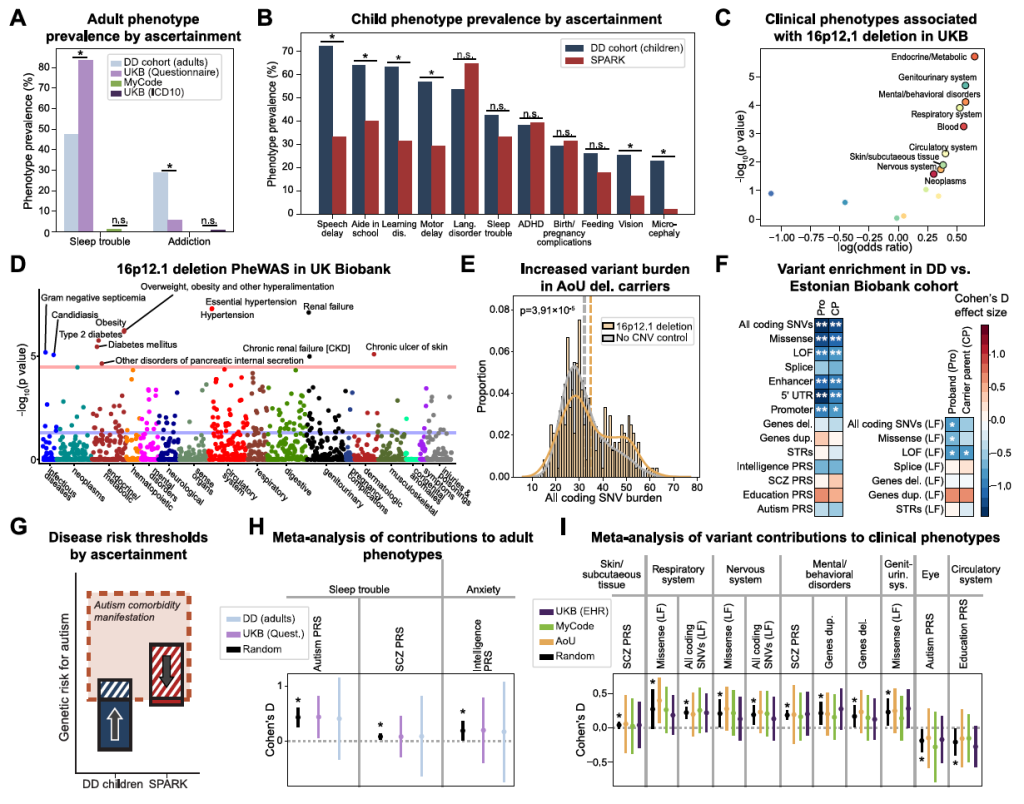
Figure S6. Effects of ascertainment on phenotypes and secondary variant associations in 16p12.1 del. carriers, related to Figure 6
2)修饰变异体与 PRS 的 “反向趋势”
UKB 队列:16p12.1 缺失携带者的错义变异负荷比健康对照低(p=0.008),且各疾病相关 PRS 均处于人群均值附近,“低风险遗传背景” 缓冲主变异风险;
AoU 队列:携带者的 SNV 负荷比对照高(p=3.91×10⁻⁵)——精神分裂症、自闭症相关 PRS 高于人群均值,“高风险遗传背景” 叠加主变异致疾病;
爱沙尼亚生物样本库(健康偏向):携带者的错义、增强子变异负荷均低于 DD 队列先证者(p≤0.009),PRS 也处于较低水平,进一步验证 “健康携带者需更低遗传负荷”。
3)关联 “反转” 实例:变异 – 表型关联的队列特异性
DD 队列儿童:修饰重复变异与喂养困难正相关(p=0.028),且喂养困难儿童的自闭症 PRS 较高(p=0.03);SPARK 队列(自闭症偏向):相同重复变异与喂养困难负相关(p=0.016),因该队列自闭症 PRS 已处于较高水平,额外变异反而产生拮抗效应。
DD 队列 ADHD 个体自闭症 PRS 升高(p=0.017),SPARK 队列 ADHD 个体自闭症 PRS 降低(p=0.035),反映不同队列中 PRS 与表型关联的适应性差异。
4
第四步:其他主变异验证 —— 修饰变异体作用 “主变异特异性”
“主变异特异性” 指次级 / 修饰变异体(含多基因风险评分 PRS)的效应并非固定,而是依赖于个体携带的 “主变异类型”—— 不同主变异(如 16p11.2 缺失、CHD8 SNV)背景下,相同次级 / 修饰变异体的作用方向、强度可能完全不同。
1)主变异类型16p11.2 缺失 / 重复
16p11.2 缺失携带者:
—— PRS 效应:精神分裂症 PRS 升高与 “全量表 IQ 升高” 呈正相关(β=0.327,p=0.031)—— 即该主变异背景下,精神分裂症相关常见变异的累积风险越高,反而 IQ 越高;
—— 次级 / 修饰变异体效应:额外携带的 “修饰缺失变异”(非 16p11.2 的其他片段缺失)与 “全量表 IQ 降低” 呈负相关(β=-0.288,p=0.039)—— 相同主变异下,不同修饰因素(PRS vs 修饰缺失)的效应甚至相反。
16p11.2 重复携带者:
—— 次级 / 修饰变异体效应:额外携带的 “修饰-缺失” 或 “修饰-重复”,均与 “自闭症行为评分降低” 呈负相关(β=-0.455~-0.522,p≤0.035)—— 即这类修饰变异体反而会减轻自闭症症状;
—— PRS 效应:未观察到任何 PRS(如自闭症 PRS、精神分裂症 PRS)与表型的显著关联—— 与 “16p11.2 缺失” 主变异下 PRS 的明显作用形成鲜明对比。
特异性体现:
同一染色体区域的 “缺失” 与 “重复” 仅为拷贝数差异(主变异类型不同),却导致次级 / 修饰变异体的作用方向(减轻 vs 降低 IQ)、PRS 的作用与否(有效应 vs 无效应)完全不同。
2)CHD8 等 SNV(神经发育强相关基因)
CHD8 是神经发育的核心调控基因,其单核苷酸变异(SNV,如错义、无义突变)属于另一类主变异,该背景下次级 / 修饰变异体与 PRS 的效应同样具有特异性。
次级 / 修饰变异体效应:
——额外携带的 “LF 缺失变异”(功能缺失型片段缺失)与 “全量表 IQ 降低” 呈负相关(β=-0.157,p=4.01×10⁻⁵)—— 效应极强且显著;
——额外携带的 “STR 扩增”(短串联重复序列异常扩增)与 “全量表 IQ 升高” 呈正相关(β=0.109,p=0.005)—— 与 “LF 缺失” 的效应完全相反。
PRS 协同效应:
——智力相关 PRS 每降低 0.1 个标准差(即智力遗传风险每升高一点),会导致 IQ 在 “LF 缺失” 的负效应基础上额外降低 2.3 分(p=0.002)—— 说明 PRS 并非独立作用,而是与次级变异体协同,共同放大对表型的影响,且这种协同仅存在于 CHD8 SNV 主变异背景下。
特异性体现:
相较于 “16p11.2 缺失” 主变异下 “PRS 与次级缺失效应相反”,CHD8 SNV 主变异下 “PRS 与次级缺失效应协同”,且 “STR 扩增” 的作用方向(升高 IQ)与其他主变异背景下(如 16p12.1 缺失中 STR 关联神经缺陷)完全不同。
3)对照验证:无主变异个体 —— 主变异是次级 / 修饰变异体发挥作用的前提
为进一步验证 “主变异特异性”,研究分析了 1528 名无任何明确致病性主变异的自闭症先证者,结果仅发现 1 个微弱关联:仅 “次级 / 修饰重复(LF)” 与 “BMI 降低” 呈负相关(β=-0.086,p=0.032)—— 无其他任何次级 / 修饰变异体(如 SNV、STR)或 PRS 与表型的显著关联。
关键结论:无主变异时,次级 / 修饰变异体的效应几乎可忽略;只有存在主变异时,次级 / 修饰变异体才能显著影响表型 —— 反证 “主变异是次级 / 修饰变异体发挥作用的必要背景,其类型直接决定修饰因素的效应是否存在”。
4)核心机制:主 – 修饰变异的交互作用—— 表型异质性的 “特异性驱动”
研究共识别出 21 个关键交互作用实例,进一步证实 “主变异特异性” 是表型差异的核心原因:
交互实例分类:
——主变异 – 次级 / 修饰变异交互(13 个):如 “CHD8 SNV 主变异” 与 “修饰-缺失变异” 对 “全量表 IQ” 的交互作用(β=-0.052,p=0.005,FDR=0.033)—— 即 “修饰-缺失” 对 IQ 的降低效应,仅在携带 CHD8 SNV 时才显著,无该主变异则无效应;
——主变异 – PRS 交互(8 个):如 “某神经发育基因 SNV 主变异” 与 “自闭症 PRS” 对 “SRS 评分” 的交互作用(β=0.064,p=0.002,FDR=0.011)—— 即 “自闭症 PRS 升高” 对社交障碍的加剧效应,仅在携带该 SNV 主变异时才出现。
交互作用的 “特异性特征”:
不同主变异与同一修饰因素的交互效应完全不同 —— 例如 “16p12.1 缺失” 主变异与 “修饰-缺失” 的交互会加剧智力障碍(β=-0.071,p=0.003),而 “16p11.2 缺失” 主变异与 “修饰-缺失” 的交互仅轻微降低 IQ(β=-0.028,p=0.049),效应强度差异达 2.5 倍。
核心价值:这类 “主变异特异性的交互作用”,正是导致 “相同主变异携带者表型差异巨大”的根本原因。
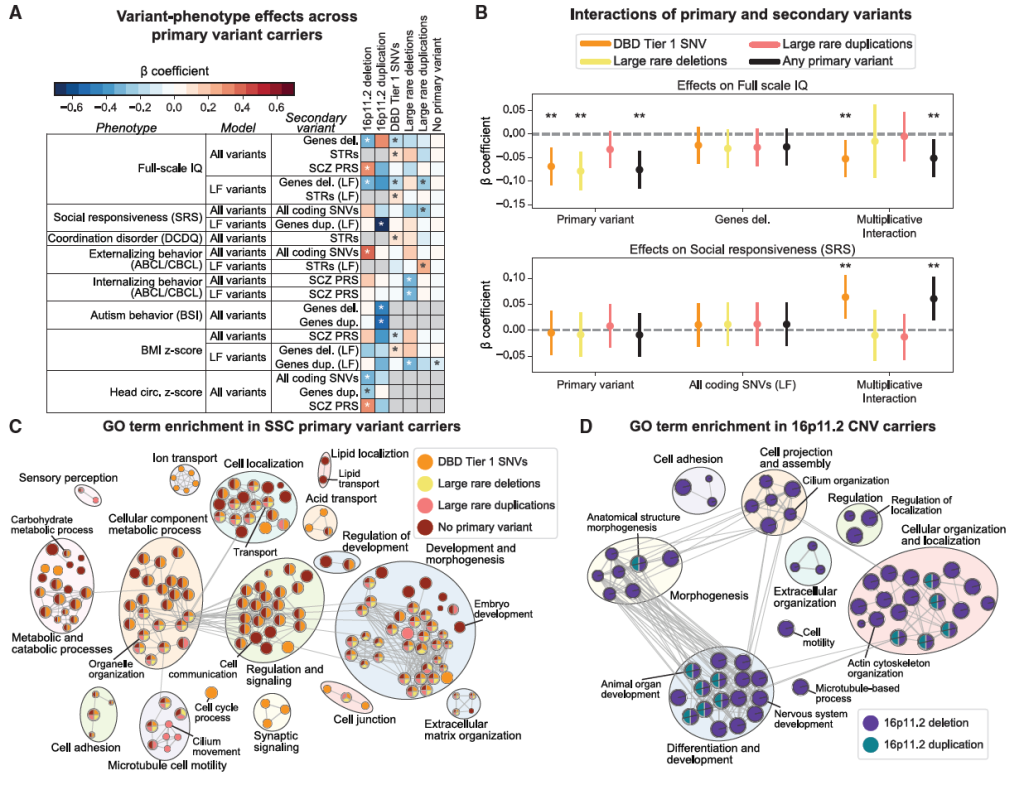
Figure 7. Secondary variant associations in probands with primary variants
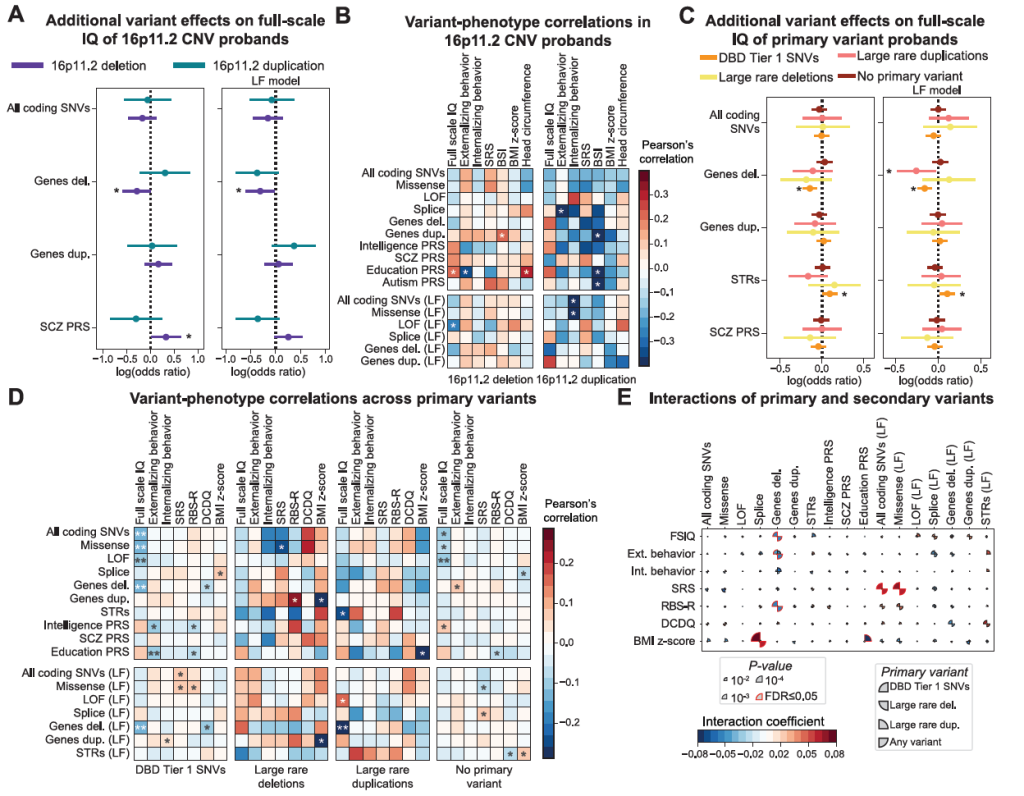
Figure S7. Associations between secondary variants and developmental features of 16p11.2 CNV probands, related to Figure 7
四
研究结论与意义:从 “科研发现” 到 “临床应用”
1
核心结论:多因素协同驱动表型异质性
修饰变异体与 PRS 的修饰效应具有 “四特异性”:主变异特异性(16p12.1 vs 16p11.2)、变异类型特异性(STR 关联神经症状、SNV 关联生长特征)、表型特异性(Hedgehog 通路变异关联行为异常)、验证方式特异性(健康 vs 疾病队列);
基因网络连接性差异(16p12.1 “分散” vs 16p11.2 “集中”)是表型异质性的关键机制,PRS 通过与不同网络的交互,进一步影响异质性程度;
验证偏倚是基因型 – 表型关联研究的重要干扰因素,跨队列荟萃分析可识别稳健关联(如智力 PRS 与 ADHD 相关,p=0.048);
主变异与修饰因素(修饰变异体、PRS)的交互作用,是表型异质性的核心驱动。
2
科学与临床意义:与既往研究的联系
1)科研范式突破
拓展修饰变异体定义:首次纳入非编码变异、STR 扩增,打破 “仅编码变异致病” 的局限,完善 “多因素协同致病” 模型;
明确 PRS 的修饰价值:证明多基因风险可 “改变” 主变异效应(如 16p11.2 缺失者的 BMI,16p12.1 缺失者的精神疾病风险);
发现跨世代累积:选型婚配(16p12.1 携带者配偶精神疾病相关性高)导致变异负荷与 PRS 风险逐代升高。
2)临床价值
遗传咨询:不能仅以 “携带主变异” 判断患病风险,需结合修饰变异体负荷 + PRS(如 UKB 中 16p12.1 携带者虽有主变异,但无修饰打击,风险低);
精准治疗:同一疾病(如自闭症)需靶向 “被扰乱的通路” 而非 “单个基因”——16p12.1 缺失者可能因修饰变异体与 PRS 的差异,分别需干预线粒体功能或突触信号;
研究设计:必须纳入多来源队列,同时关注次级变异体与 PRS,避免 “仅选患者” 导致的偏倚(如仅研究 DD 队列会高估神经症状发生率)。
3
研究局限性
1)统计效力:部分关联仅达 “名义显著性”(如 FDR=0.586),需更大队列验证;
2)技术限制:短读长测序对 STR、复杂结构变异的检测准确性不足,PRS 计算依赖参考人群,可能存在人群分层偏差;
3)队列差异:不同队列基因分型方法不同(如 WGS vs 微阵列),PRS 计算参数存在差异,无法直接对比变异负荷。
五
总结
该研究发现修饰变异体对 16p12.1 缺失及其他主变异体外显度的影响,存在 “家系特异性、表型特异性、验证方式特异性及主变异体特异性” 模式。
16p12.1 缺失及其他主变异体的复杂性,呼吁临床采用 “个体化医疗” 策略 —— 在遗传咨询、疾病管理及治疗方案制定中,充分考虑个体水平的表型特征与更全面的基因组变异类型检测。
参考文献:
Matthew Jensen, et al. Genetic modifiers and ascertainment drive variable expressivity of complex disorders. 2025, Cell 188, 1–18. https://doi.org/10.1016/j.cell.2025.09.012