综述| 非翻译区域变异在孟德尔疾病中的作用(下)
- boke
- 2025-08-19
- 9:40 上午
非翻译区(UTRs)位于基因的蛋白质编码序列两侧。5′ UTR和3′ UTR序列通过线性结构和结构元件介导转录后调控,控制RNA的稳定性、细胞定位和蛋白质翻译速率。5′ 和3′ UTR内的变异已被证明可以通过多种不同的机制导致疾病。
然而,为了使这些变异能够常规地注释和解释在临床遗传检测中,我们需要更好地了解这些区域及其中的致病变异谱。在本综述中,我们系统地评估了先前在UTRs中识别出的孟德尔致病变异,并列出了其潜在机制。随着基因组测序的广泛应用和越来越多地应用于诊断环境,本综述将为考虑和解释UTR变异提供宝贵资源。
5′ UTR重叠启动子的变异干扰转录起始
启动子是DNA中启动基因的转录的序列。相关的蛋白质,称为转录因子,它们与启动子基序结合来启动转录。启动子通常跨越基因的TSS,这也标志着5′ UTR的开始。因此,启动子下游的部分也可能与5′ UTR重叠。
在两个无关家系中观察到的BRCA1的5′ UTR-重叠启动子区域的变异(NM_007294.4:c.107A > T)与通过等位基因特异性启动子甲基化导致的表观遗传沉默相关,并认为可能导致3级乳腺癌或高等级卵巢癌。然而,另一项研究对此却提出了质疑:作者在更大规模的 BRCA1 等位基因特异性启动子甲基化患者队列中寻找这种变异,但没有发现这种变异。另一个例子是报告重叠MERTK基因的TSS变异(NM_006343.3:c.125G > A),被假设破坏转录,改变二级结构并导致遗传性视网膜病。并在体外实验中发现mRNA水平降低,支持这一猜想。
变异可通过对mRNA二级结构造成破坏而引发疾病
mRNA是一种单链RNA序列,能够通过自身互补碱基配对折叠并形成二级结构。这些结构可以通过干扰核糖体扫描效率和降低mRNA稳定性来影响翻译。当mRNA链折叠并与相邻部分配对时,存在几种类型的二级结构,包括pseudoknots、G-四联体和发夹结构(也称为茎环)。仅根据序列预测mRNA的确切构象和结构是困难的,并且二级结构通常是动态的。
铁响应元件(IRE)是研究最深入的茎环结构之一,它影响铁稳态相关的mRNA的翻译。细胞对铁的摄取必须严格调控,因为铁的不足或过量都可能造成损害。mRNA的5′ 帽附近的一个保守的IRE茎环被铁调节蛋白1或2(IRP1/IRP2)结合。IRP的结合通过阻止核糖体进入5′ UTR来抑制翻译起始。IRPs通过直接与细胞质中的铁相互作用来调节铁的可用性。L-铁蛋白结合并储存细胞中的铁。高铁蛋白血症/白内障综合征(HHCS)是由L-铁蛋白(FTL)基因5′ UTR中的IRE突变(例如NM_000146.4:c.- 164C > T)引起的,该变异阻止了其与IRPs的相互作用,导致L-铁蛋白产生水平失调性升高。
另一个涉及改变mRNA二级结构的例子是一个位于ADAR1基因的5′ UTR中的杂合子变异(NM_001111.5:c.60A > G),该变异降低基因表达,并被报道可引起对称性遗传性色素沉着病。这个变异似乎不会破坏该区域已知的任何调控特征;然而,这种单核苷酸变化被认为足以改变mRNA的结构。这种结构变化如何导致基因表达降低目前尚不清楚。
SEPN1相关肌病包括四种常染色体隐性遗传病。SEPN1产生硒蛋白,这对于正常肌肉发育是必需的。所有硒蛋白的一个独特特征是存在氨基酸硒半胱氨酸,它非常规的由CDS终止密码子UGA所编码。这种UGA的重新编码是通过一个高度保守的茎环结构实现的,该结构从CDS UGA密码子下游6个碱基处开始,位于3′ UTR中。这个区域被称为硒半胱氨酸插入序列(SECIS)。一个SECIS RBP(SBP2),它结合SECIS,对于将UGA重新定义为硒半胱氨酸密码子至关重要,防止在UGA处终止。在UGA处终止会触发NMD并导致蛋白质不足。有3种变异(例如NM_020451.3:c.*1107T > C)与SEPN1相关肌病有关,因为它们干扰SBP2的结合,显著降低mRNA和蛋白质水平。
5′ UTR变异可以改变CDS起始密码子的翻译起始效率
精确的CDS起始密码子周围的上下游序列决定了从AUG翻译的强度以及产生的蛋白质量。5’UTR中的变异,如果破坏了Krizak共识序列,可能会影响疾病风险。Nicole等人最近的一篇论文描述了RARS2(NM_020320.5:c.2T > C)5′ UTR中的一个变异,该变异改变了CDS起始密码子的Kozak序列,减少了蛋白质产量并导致小脑发育不良(PCH)。作者还评估了ClinVar中其他预测会改变CDS Kozak序列的变异,发现有20个,其中大部分被标记为未知意义的变异。他们得出结论,这类变异可能是被低估的疾病机制。
变异可影响3′ UTR的聚腺苷酸化
对于大多数编码蛋白质的基因,pre-mRNA的3′ 端通过转录过程中的连续切割和加poly(A)反应形成。这是一个广泛研究的现象,由Curiinha等人进行综述,并在本文中总结。3′ UTRs含有一个加poly(A)位点(PAS),它指导在转录的后期阶段添加数百个腺嘌呤残基,形成polyA尾巴。polyA尾巴在mRNA的出核、翻译和稳定性中起着重要作用。特定PAS在pre-mRNA中的使用受RNA顺式元件和多个转录因子调控。最重要的顺式元件是polyA信号,一个位于PAS上游约~10-35碱基对的六聚体(通常是AUAAAA或其类似物如UAUAAA)。
多种影响多聚腺苷酸化的变异与疾病相关。例如,NAA10基因的PAS区(例如NM_003491.4:c. *43A>G) 内的几个变异已被与X连锁的microphthalmia综合征相关。体外研究表明,这些变异会破坏切割和多聚腺苷酸化,导致mRNA水平降低。相反,F2基因 3′ UTR末端的一个功能获得性单核苷酸变异(NM_000506.5:c. *97G>A )会导致凝血酶原血浆水平升高。野生型多聚腺苷酸化切割信号效率低下,但变异增加了切割位点识别、增加了3′ 端处理、mRNA积累和蛋白质合成,导致血栓形成。
尽管不是严格的3′ UTR变异体,但一个改变α珠蛋白(HBA1)CDS终止密码子从UAA到CAA(NM_000558.5:c.427T > C)的单核苷酸变异体,允许翻译核糖体进入3′ UTR。这与α珠蛋白mRNA半衰期的显著降低有关。进一步的实验阐明,存在富含α的区域与α复合物相互作用,该复合物包含防止mRNA降解的蛋白质。α复合物被认为可以保护poly(A)尾巴并稳定mRNA。如果这种相互作用被阻止,例如在本例中,poly(A)尾巴会加速缩短;mRNA会过早降解,从而导致α地中海贫血。
变异破microRNA及其结合位点,改变RNA沉默
miRNAs是非编码RNA,长度约为22个碱基,包含一个或多个发夹环。它们参与RNA沉默和基因表达的转录后调控。miRNA与mRNA上的互补序列配对,通常在3′ UTR内,可以沉默并抑制蛋白质的产生,例如通过mRNA去腺苷化和去帽化。
存在多个miRNA内部的致病性变异,特别是在负责介导mRNA结合的“种子区域”内。此外,3′ UTR内的miRNA结合位点变异可能会破坏miRNA介导的调控。例如,COL4A1基因3′ UTR中一个7碱基区域的变异(例如NM_001845.6:c.*31G > T)消除了miR-29 miRNA的结合位点,导致COL4A1 mRNA水平升高,进而引发脑小血管病。相反,REEP1基因 3′ UTR中miR-140结合位点的两个变异(例如NM_001371279.1:c.808C > T)预测会抑制miRNA对翻译的影响,导致REEP1蛋白水平降低,导致遗传性痉挛性截瘫31型。
讨论
在此,我们系统地回顾了5′ 和3′ UTR中重要的调控元件,这些元件在受到扰动时可能导致孟德尔疾病。这些区域在历史上一直被忽视,但随着基因组测序在临床诊断测序中的发展和应用,而现在UTR区域的序列信息正变得更加容易获取。近期关于临床解释非编码变异的建议有助于在临床环境中对UTR变异进行分类;不过,注释和解释这些变异仍然是一个相当大的挑战。
注释UTR内的变体目前需要使用一系列生物信息学工具和数据集。每个工具可以根据特定的假设效应注释变体,例如,UTRannotator,但并没有单一方案能够结合所有已知的调控元件和变异机制。每个变异可能也有多个预测效果,例如,创建一个uAUG并改变转录调控因子结合位点。在没有广泛的功能性表征的情况下,确定一个变异如何调节蛋白质表达的效果可能会很困难。
目前,UTR变体通常根据已知的调控元件进行注释;然而,这些元件可能注释不完整,尤其是如果它们是时空特异性的。此外,可能还存在通过变异介导其效应的、尚未知的调控元件类别。这通过据报道导致疾病的UTR变异得到强调,这些变异通过未知机制起作用。一个例子是VMA21基因的3′ UTR中96个碱基的缺失,它与X连锁过度自噬性肌病(XMEA)相关联,并已被证明可以减少mRNA数量。其潜在机制尚不清楚,但可能涉及mRNA的降解。
解读UTR变异体,尤其是非编码变异体的关键挑战在于,它们通常具有不完整或低度表型的效应。此外,这些效应可能朝两个方向,导致蛋白质水平增加或减少,如上文中详细描述的许多变异体所示。例如,MEF2C中的“uoORF创建”变异会降低蛋白质表达,而THPO 5′ UTR中的变异会增加蛋白质表达,两者均导致相应的疾病。蛋白质表达增加或减少导致疾病阈值高度基因特异性,但这些阈值对于大多数基因而言是未知的。对于引起疾病的部分效应非编码变异体,它们可能导致较轻的表型,例如,KLHL40 CDS中的变异与严重的肌萎缩侧索硬化症相关,而KLHL40 5′ UTR中的变异与较轻的疾病形式相关。此外,引起疾病的部分效应非编码变异还可能导致疾病发病较晚和/或降低疾病外显率。
UTR变异的解释也因需要考虑更广泛的序列背景而变得复杂。UTR是具有精确调节元素组合的复杂调节元件。与观察到的增强子中的冗余相似,其他调节元件如果其中一个元件被基因变异破坏,也可能起到补偿作用。例如,在miRNA结合位点,可以在同一3′ UTR内发现同一miRNA的多个结合位点。在解读5′ UTR变异对翻译调节的影响时,考虑更广泛的序列背景同样至关重要。例如,并非所有上游起始密码子创建的变异都是相同的:从上游起始的翻译可能性不仅取决于与Kozak共识的周围序列匹配,还取决于其他翻译的uORF的位置。例如,如果起始点位于广泛翻译的uORF内,它将不会被扫描的核糖体“看到”,因此不会对下游CDS翻译产生任何影响。
考虑到UTR变异解释的挑战,功能特征化非常重要。除了对患者在实验中观察到的单个变异效应进行解码的实验外,大规模多联测试变异效应在变异解释方面具有巨大潜力,并且有助于我们更深入地了解基因调控。然而,值得注意的是,UTR中的变异可以通过多种不同的机制起作用:影响转录、RNA加工、RNA稳定性和翻译。除非在实验中捕捉到所有这些机制,否则不能排除变异的影响。另外,如果一种检测侧重于下游影响,如细胞活力,作用的确切机制可能仍然不明确。此外,如上所述,UTR作为完整的功能元件,包括整个序列在其原生环境中可能是准确功能表征的关键。
5′ 和3′ UTR的变异是罕见但未被充分认识的孟德尔遗传病的病因。在临床环境中注释和解读这些变异具有挑战性,但可为患者提供诊断,从而增加我们对UTR介导的基因调控的了解。
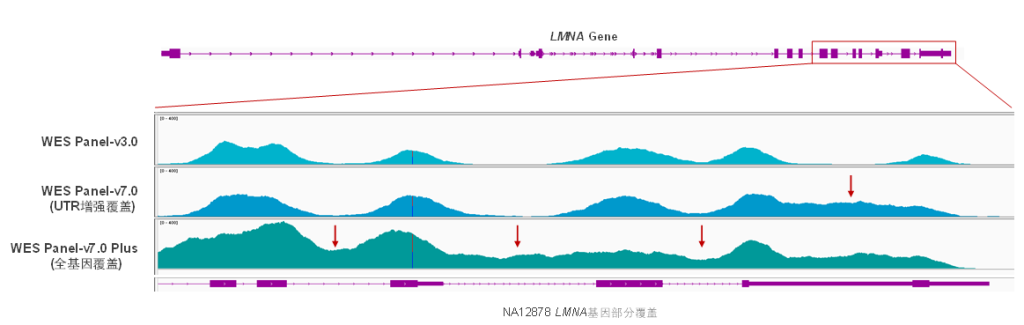
伯科设计合成的人全外显子Panel对多个基因的UTR区域进行了加强覆盖,搭配非编码区致病位点补充Panel,可以对UTR区域进行更全面的检测,保证非翻译区变异的深入研究。
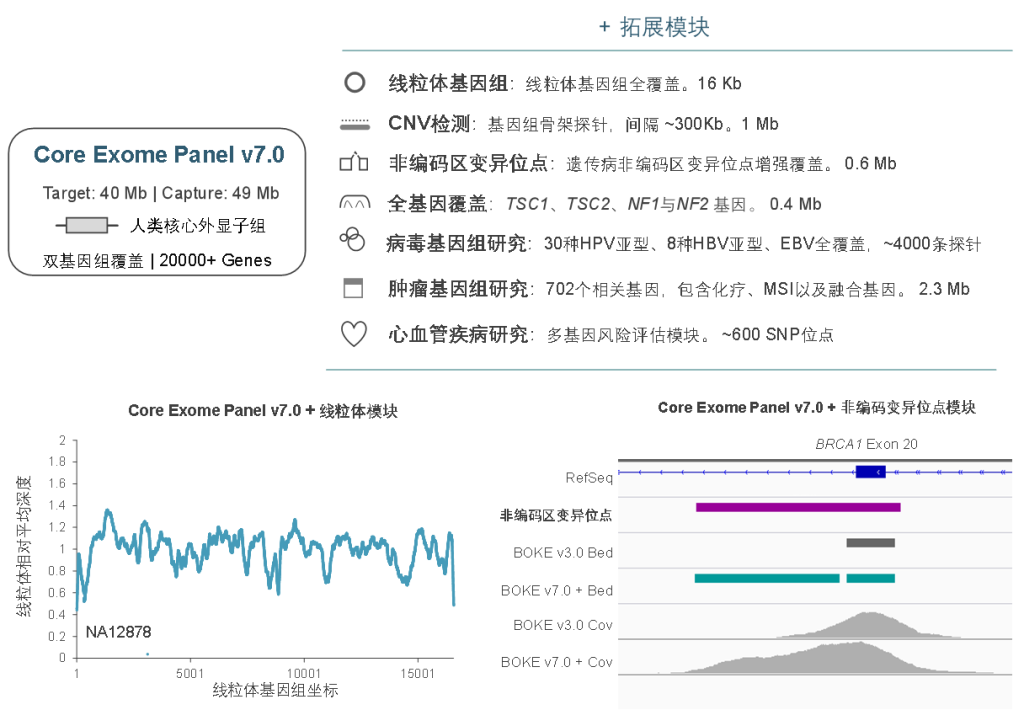
TargetCap® Core Exome Panel v7.0
TargetCap® Core Exome Panel v7.0(下文简称BOKE v7.0),该WES Panel增强了基因组hg19传统研究区域的覆盖,兼顾 hg19 & hg38 双版本基因组,可以更好的保证临床科研与转化的延续性。目标区域和捕获区域大小分别为40Mb和49Mb,对 hg19 传统研究区域覆盖提升至99.7%(友商I-v1),hg38传统研究区域覆盖相近(友商A-v8)。同时,新添加数百个具有一定功能与表型的基因,总基因数量达到20000+。
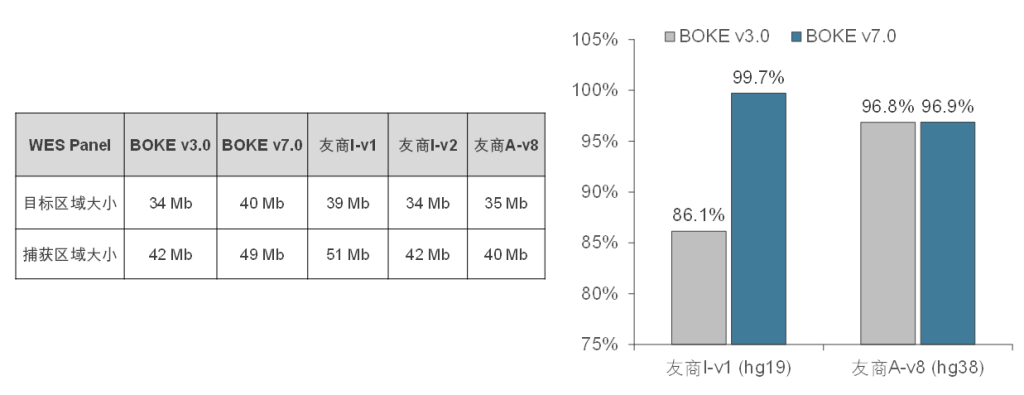
TargetCap® Core Exome Panel v7.0目标区域大小以及对不同友商产品目标区域的覆盖情况
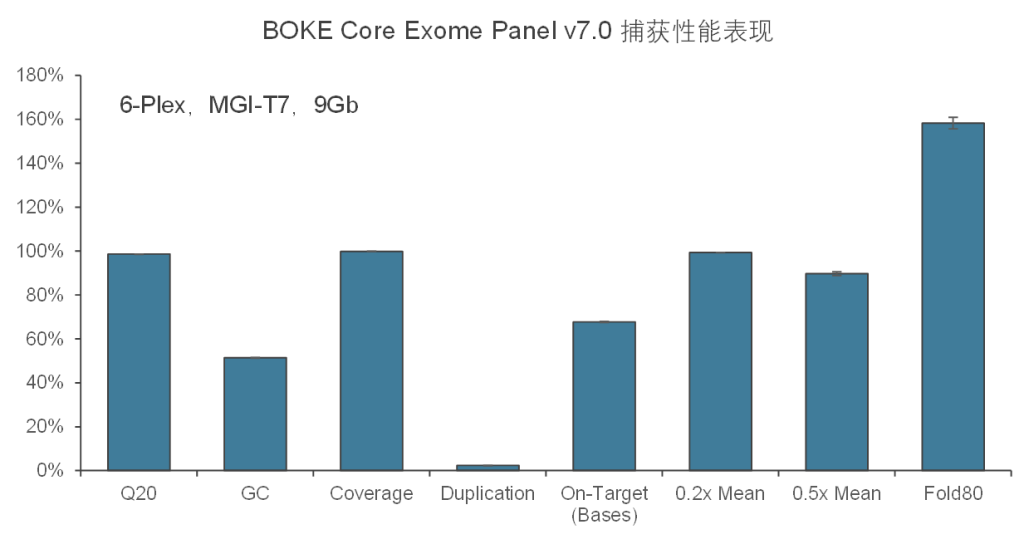
在捕获性能方面,TargetCap® Core Exome Panel v7.0依然表现优异,与TargetCap® Core Exome Panel v3.0表现相近。在测序9Gb条件下,平均深度达到110x左右,20x和30x以上区域占比分别为99.5%和98.5%,Fold 80为1.5-1.6之间,与国际领先产品数据表现相当。
同时,TargetCap® Core Exome Panel v7.0也可灵活的与拓展模块组合使用,满足不同场景的临床研究的需求及转化应用,包括线粒体、遗传病非编码区变异位点、单基因全覆盖、病毒基因组、肿瘤全景变异检测、重大疾病多基因风险评估模块等。此外,伯科公司自研自造的寡核苷酸合成平台可以快速响应个性化定制的需求,为人类基因组分子遗传学的研究与转化,提供更加全面高效的解决方案。