


人全外显子液相基因芯片(WES)临床应用的进展-心血管疾病(下)
- boke
- 2025-06-23
- 10:47 上午
心房颤动(房颤)
超5万心房颤动病例测序数据(41250 WES+超10000 WGS),揭示了心房颤动与各种遗传性心肌病之间存在共同的生物学基础。
文献标题:Sequencing in over 50,000 cases identifies coding and structural variation underlying atrial fibrillation risk
发表期刊:Nature Genetics
发表时间:2025年

背景:心房颤动 (Atrial fibrillation,AF)是最常见的持续性心律失常显著增加死亡、卒中、心力衰竭(心衰)和认知功能障碍风险,严重影响患者生活质量。
已知心房颤动具有很大的遗传成分,来自常见变异的遗传力超过23%,先前全基因组关联研究(GWAS)已鉴定出与AF相关的140多个基因组位点。
全基因组测序(WGS)和全外显子测序(WES)研究为蛋白质编码区域的分析提供了最高的分辨率,并能够发现具有大遗传效应的罕见变异。2018年,使用家族和早发性AF病例的研究发现了TTN中罕见失活变异与AF之间的关联,这一关联随后在英国生物样本库的独立分析中得到证实。尽管这些研究使我们理解了TTN中罕见变异对AF风险的贡献,但这些变异仅解释了AF易感性变异的0.2%。
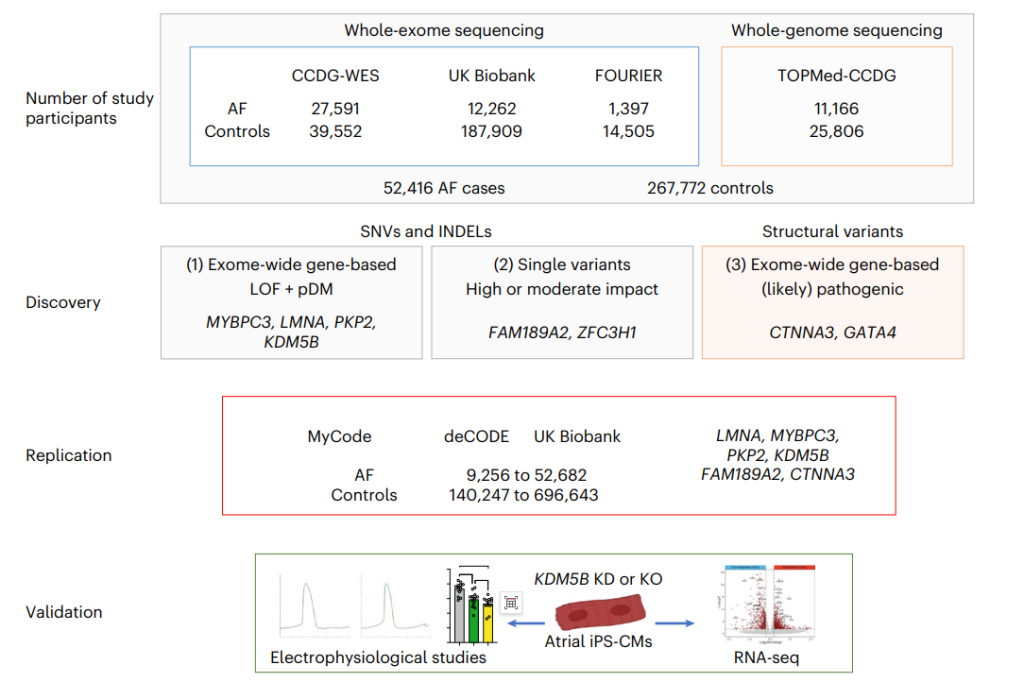
结果:通过整合分析来自CCDG、FOURIER以及UKB的近238,000个WES样本和TOPMed的超过36,000个WGS样本,对复杂疾病房颤(AF)进行了研究。
这项研究利用目前最大规模的测序数据,包括52,416例AF病例和277,762例对照。通过WES序对蛋白质编码基因进行检测,评估罕见编码遗传变异与心房颤动风险的关系。WGS数据用于识别具有高度影响的预测功能性结构变异,包括大型缺失、重复、插入和倒位。
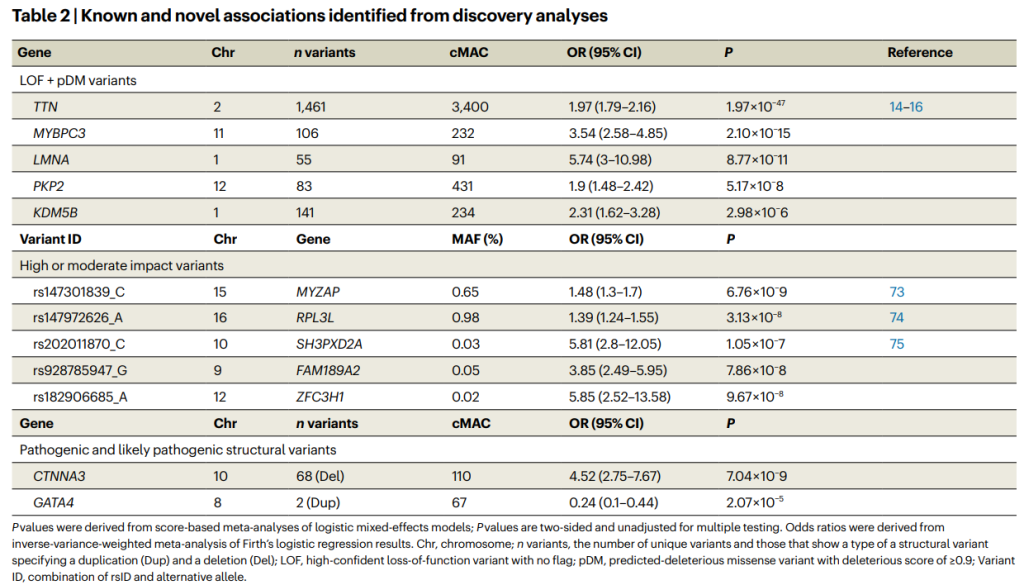
在罕见编码变异的负担测试中,发现了4个与AF相关的新基因,MYBPC3、LMNA、PKP2和KDM5B,这些基因与心肌病高度关联。MYBPC3基因变异是肥厚性心肌病的重要原因之一。LMNA 基因的致病变异与多种疾病有关,包括埃默里-德雷福斯肌营养不良症、家族性部分脂肪营养不良症、肢带肌营养不良症和扩张型心肌病。类似地,PKP2基因与阵发性右室心肌病相关。在UKB中,KDM5B基因变异与多种心血管特征相关,包括心力衰竭、左心室射血分数降低和室性心律失常。此外,在先天性心脏病病例中发现了KDM5B罕见变异的富集。
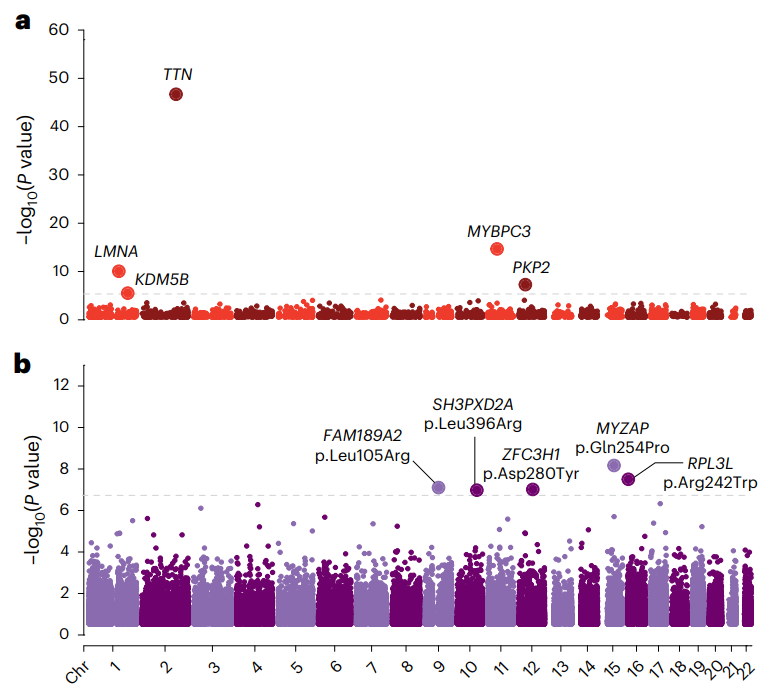
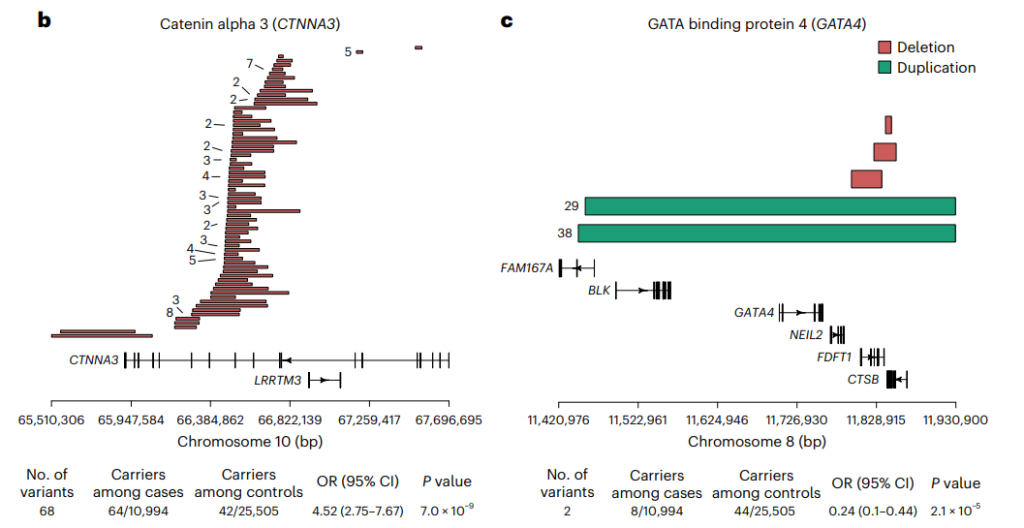
此外,还发现了AF与CTNNA3缺失和GATA4重复导致的罕见结构变异之间的关联。在数量有限的具有右心室心肌病病例中已经发现CTNNA3的罕见变异,并且Ctnna3敲除小鼠会发展成扩张型心肌病。GATA4的LOF变异是先天性心脏病的已知原因,增加GATA4剂量可能对房颤具有保护作用,但这种保护的潜在机制需要进一步研究。
该研究还在MyCode、deCODE和UK Biobank的独立样本中广泛复制了这些发现。最后,还发现敲除干细胞来源的房室心肌细胞中的KDM5B会导致动作电位持续时间缩短以及与心房稳态和传导相关的基因广泛的转录本调节失调。
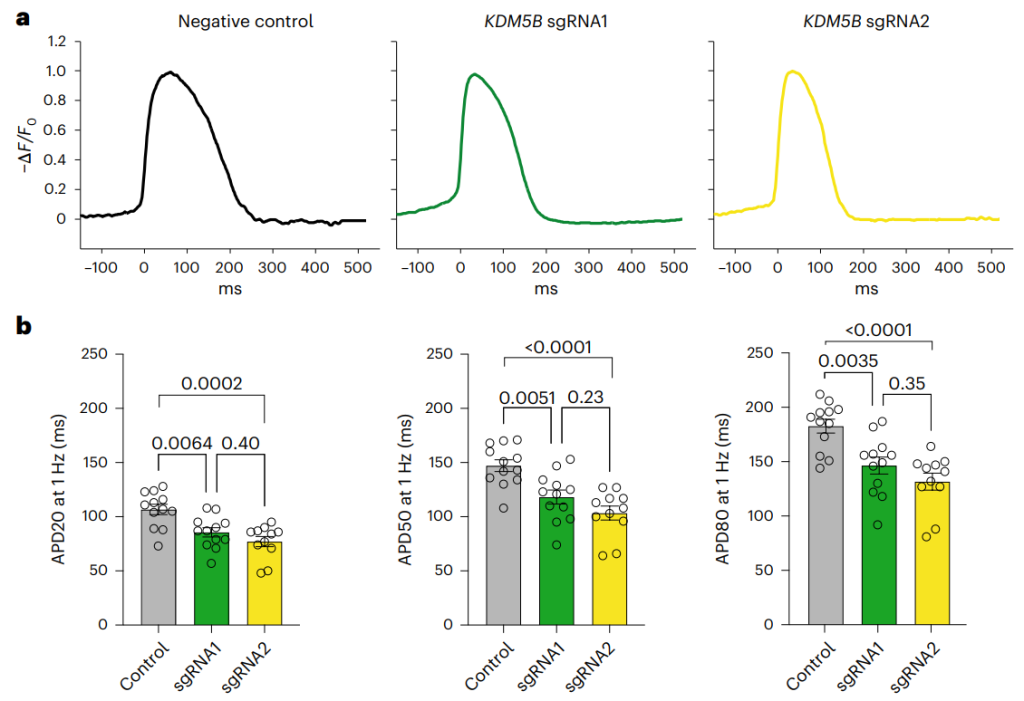
研究结果突出了罕见编码和结构变异对AF的贡献,揭示了心房颤动与各种遗传性心肌病之间存在共同的生物学基础。总的来说,这项工作强调了大规模测序在了解复杂疾病的罕见遗传机制方面的价值。
冠心病
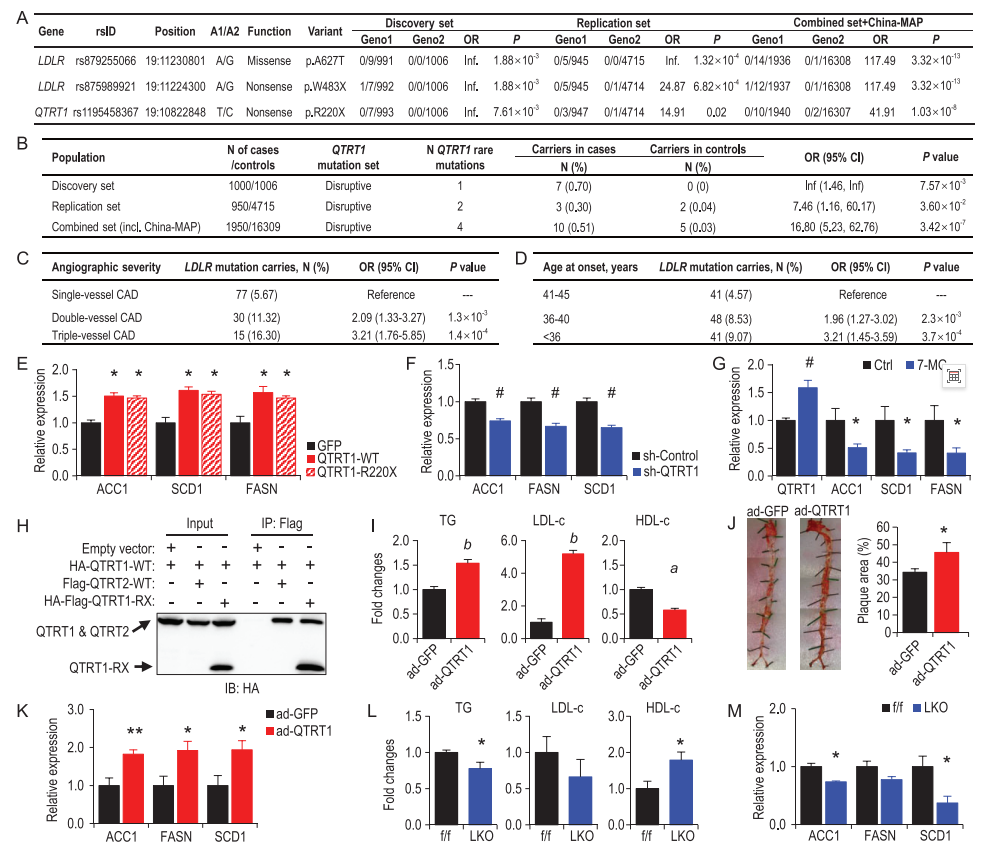
1. 一项包含7671名汉族个体的CAD全外显子测序研究,识别出LDLR和QTRT1基因中的罕见编码变异增加早发冠心病风险;进一步的机制研究表明,新致病基因QTRT1变异通过从头脂肪生成失调和加速动脉粥样硬化影响早发冠心病风险。
文献标题:Exome sequencing identifies rare mutations of LDLR and QTRT1 conferring risk for early-onset coronary artery disease in Chinese
发表期刊:National Science Review
发表时间:2022年

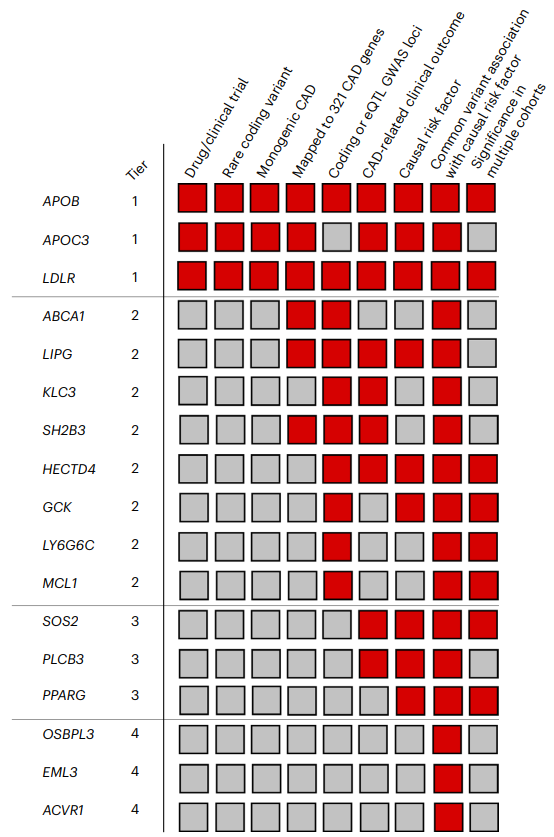
2. 机器学习定量描述冠心病表型,深度挖掘~60万人WES数据中冠心病罕见致病变异,促进冠心病致病机制研究
文献标题:Exome sequence analysis identifies rare coding variants associated with a machine learning-based marker for coronary artery disease
发表期刊:Nature Genetics
发表时间:2024年

冠状动脉疾病(CAD)是由多种风险因素和病理过程组成的一类疾病,通过简单的是否来作为疾病判定,无法准确描述CAD的进展和严重程度,因此难以发现在人群中发生频率较低的罕见基因变异。
利用机器学习对电子健康记录中的临床数据构建CAD的连续表型评价系统(ISCAD),能够发现该疾病谱上的疾病进展、严重程度和漏诊情况,可能会增强对CAD的遗传发现能力。
在本研究中,测试了来自英国生物库(UKB)、All of Us研究计划和BioMe生物库中604,915个体WES数据中的罕见和超罕见编码变异(coding variants)与冠状动脉疾病表型评分-ISCAD的关联。
研究结果显示,有17个基因发现了关联;其中,14个基因显示出至少中等程度的先验遗传、生物和/或与CAD相关的临床支持。此外,还观察到超罕见编码变异在321个CAD强相关基因中出现了聚集,提示还有更多超罕见变异的关联有待发现。这些结果扩展了我们对CAD遗传病因的理解,并说明了多标志物如何增强对复杂疾病的遗传关联研究。
背景:CHD 占所有出生缺陷的三分之一左右,全球新生儿发病率为1%至1.8%,所有年龄段患病率为0.16%。通过手术和导管介入治疗结构性心脏畸形的姑息治疗和修复,以及医疗管理的改善,使得近90%的 CHD 患者能够存活至成年,目前美国有240万人患有 CHD,其中包括140万成年人。尽管取得了显著进步,但包括心内膜炎、心律失常、再次手术、心力衰竭、肺动脉高压和神经发育缺陷在内的多种并发症或合并症使 CHD 成为一种常见的终身疾病。
CHD具有强烈的遗传基础。约25%的病例与大型染色体异常或拷贝数变异相,许多单基因变异也与 CHD 相关。隐性基因型(RGs)通常通过近亲家系和候选基因分析得到。尽管如此,对最常见的 CHD 形式遗传贡献的合理估计,这些形式通常为散发性,只有在过去十年中随着NGS测序技术的出现和 CHD 先证者及其父母样本的无偏收集才成为可能。
结果:我们分析了来自NHLBI Bench-to-Bassinet项目的5,424例 CHD 先证者的全外显子测序结果。罕见的破坏性的隐性基因型(RGs)估计至少导致2.2%的 CHD,相比于其他亚组(1.4%),其在偏侧表型中更多富集(5.4%)。
在108个经过筛选的人类隐性 CHD 基因中,发现了66个RGs,位于23个基因中,而85个基因没有RG。其中54 RGs个位于11个的基因中(有>1个RG),剩余12个基因有1个RG。近亲家系后代的RGs(4.7%,32/675)比在非近亲先证者(0.7%,34/4749)更为普遍。在410名阿什肯纳兹犹太先证者中,GDF1 和 PLD1 中的创始变异占RGs贡献的 74 %。
我们鉴定出 C1orf127 基因中存在广泛的RGs富集,该基因编码一种可能分泌的蛋白,在胚胎小鼠的脊索中表达,并与偏侧缺陷相关。
TargetCap® Core Exome Panel v3.0
TargetCap@ Core Exome Panel v3.0基于伯科高品质DNA探针合成技术开发,全流程国产制造,由~40万条探针组成,以GRCh38/hg38人类参考基因组设计,参考Refseq、CCDS、ClinVar等数据库,覆盖19,524个基因,目标区域为33.9Mb。
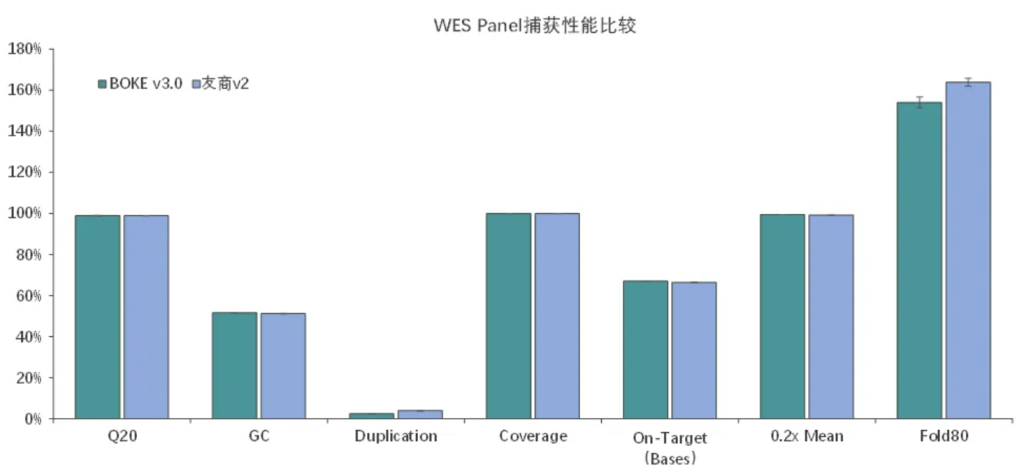
捕获性能比较
伯科全外芯片v3.0性能优异与国外友商同类型产品v2相当,中靶率、覆盖率、覆盖均一性等参数均达到国际领先水平。
适配高通量流程平台
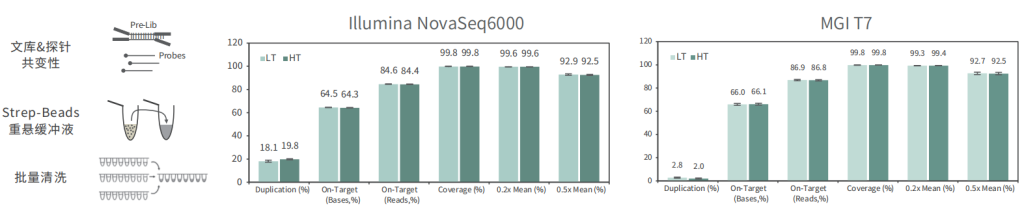
批次稳定
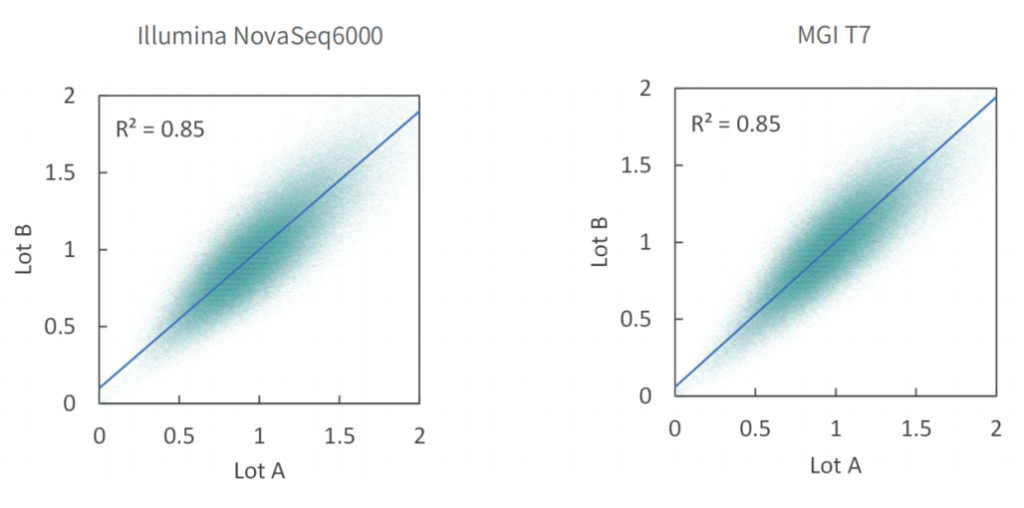
使用不同批次TargetCap® Core Exome Panel v3.0芯片对NA12878 gDNA进行捕获测序,结果显示,不同批次芯片在不同测序平台上均显示出优异的稳定性,不同位点的相对深度相关性高,批次稳定。
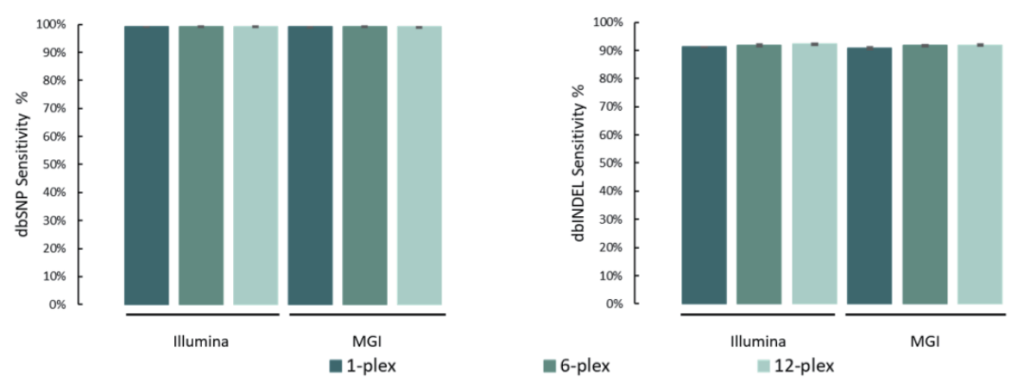
变异检测准确
单核苷酸变异(SNV)和插入缺失 (INDEL)是基因组变异的常见形式,也是引起人类疾病的重要原因。
选取NA12878标准品,与预期SNV和INDEL变异进行比较。结果表明,在MGI与Illumina测序平台,SNP灵敏度为99.1%,INDEL灵敏度为91.6%。
添加线粒体模块临床样本表现
20例全血样本(S1#-S20#),采用1-4 Plex方式使用伯科Core Exome Panel v3.0添加线粒体模块进行过夜杂交捕获;其中,S1#-S10#在Illumina NovaSeq X Plus平台测序, S11#-S20#在MGI MGISEQ-T7平台测序,均采用150PE模式测序。得到测序数据后,抽取8Gb数据进行生信分析。
两种测序平台的数据表现相近,平均深度分别为111x/115x (Illumina/MGI),中靶率优异均> 85%,覆盖均一性极佳(0.2X_MD≥99.3%);仅使用8Gb数据,高达98.5%的捕获区域达到了30X以上,99.5%的捕获区域达到20X以上,为临床样本检测提供了可靠的捕获数据。
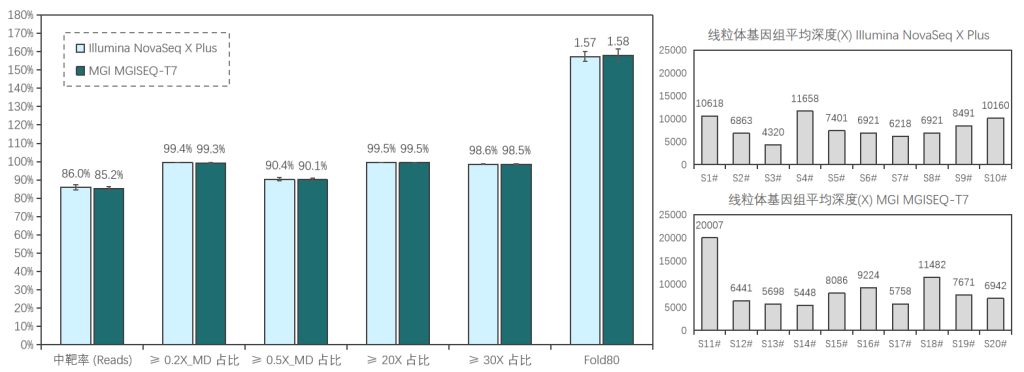
≥0.2X/0.5X_MD: Mean Depth,覆盖深度≥平均深度的0.2/0.5倍深度的区域占总区域的比例,用于表征覆盖均一性性能,越接近100%越好。
TargetCap® Core Exome Panel v7.0
TargetCap® Core Exome Panel v7.0(下文简称BOKE v7.0),该WES Panel增强了基因组hg19传统研究区域的覆盖,兼顾 hg19 & hg38 双版本基因组,可以更好的保证临床科研与转化的延续性。目标区域和捕获区域大小分别为40Mb和49Mb,对 hg19 传统研究区域覆盖提升至99.7%(友商I-v1),hg38传统研究区域覆盖相近(友商A-v8)。同时,新添加数百个具有一定功能与表型的基因,总基因数量达到20000+。
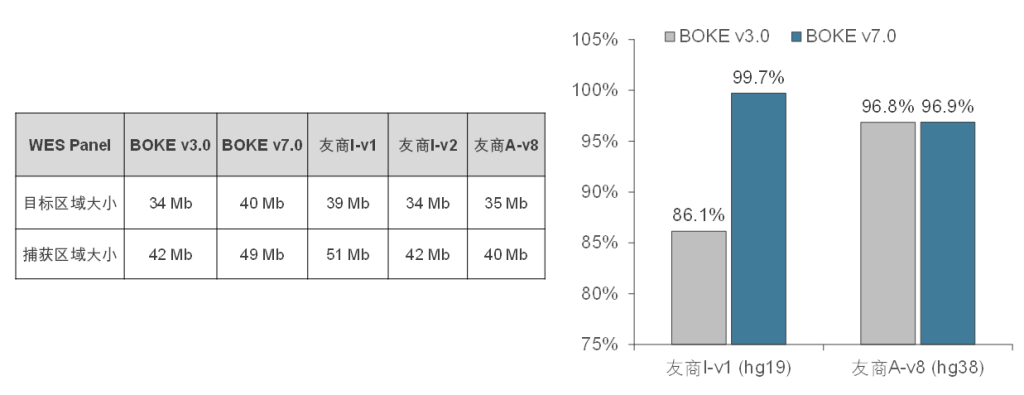
TargetCap® Core Exome Panel v7.0目标区域大小以及对不同友商产品目标区域的覆盖情况
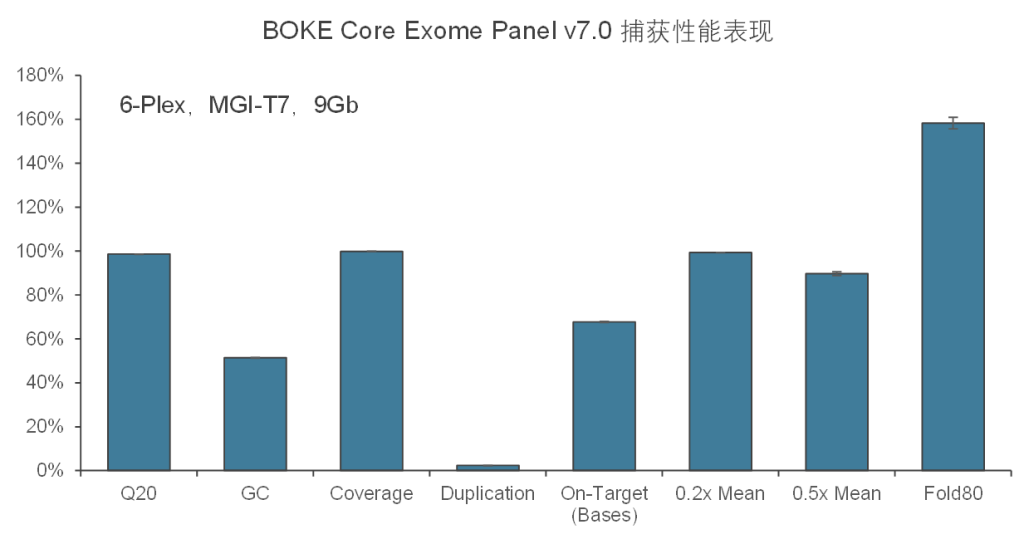
在捕获性能方面,TargetCap® Core Exome Panel v7.0依然表现优异,与TargetCap® Core Exome Panel v3.0表现相近。在测序9Gb条件下,平均深度达到110x左右,20x和30x以上区域占比分别为99.5%和98.5%,Fold 80为1.5-1.6之间,与国际领先产品数据表现相当。
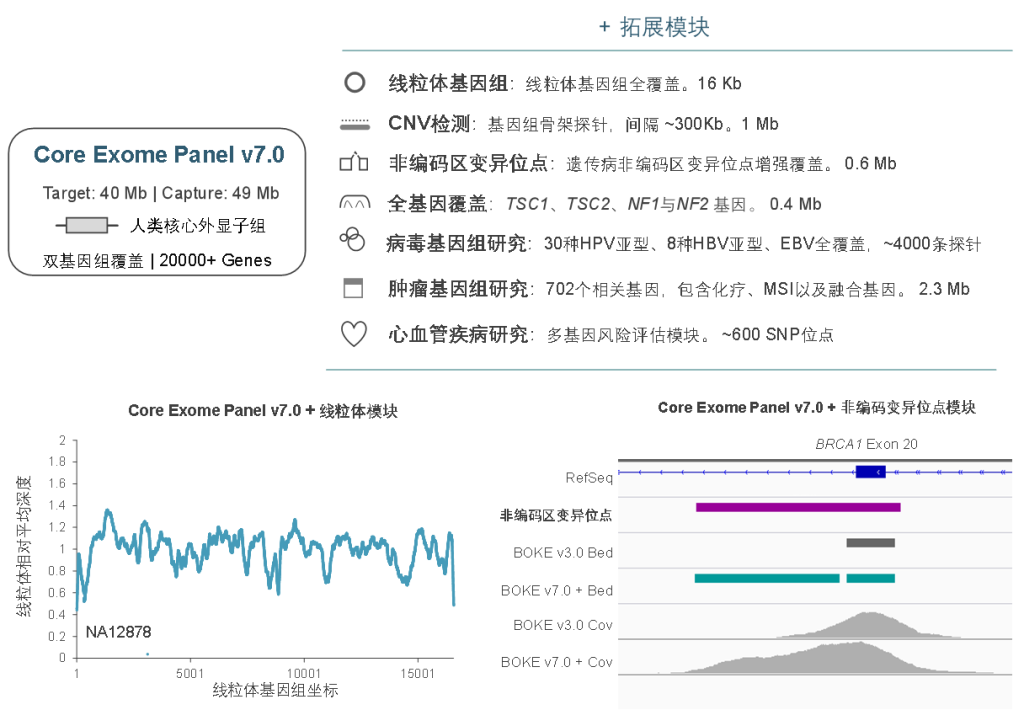
同时,TargetCap® Core Exome Panel v7.0也可灵活的与拓展模块组合使用,满足不同场景的临床研究的需求及转化应用,包括线粒体、遗传病非编码区变异位点、单基因全覆盖、病毒基因组、肿瘤全景变异检测、重大疾病多基因风险评估模块等。此外,伯科公司自研自造的寡核苷酸合成平台可以快速响应个性化定制的需求,为人类基因组分子遗传学的研究与转化,提供更加全面高效的解决方案。
参考资料
1. Choi SH, Jurgens SJ, Xiao L, et al. Sequencing in over 50,000 cases identifies coding and structural variation underlying atrial fibrillation risk. Nat Genet. 2025;57(3):548-562.
2. Yao K, Dai Y, Shen J, et al. Exome sequencing identifies rare mutations of LDLR and QTRT1 conferring risk for early-onset coronary artery disease in Chinese. Natl Sci Rev. 2022;9(8):nwac102. Published 2022 May 31.
3. Petrazzini B O , Forrest I S , Rocheleau G ,et al.Exome sequence analysis identifies rare coding variants associated with a machine learning-based marker for coronary artery disease[J].Nature Genetics, 2024, 56(7):1412-1419.