


2023NRG综述|回顾孟德尔遗传学:医学遗传学中的显性与隐性(一)
- boke
- 2023-04-25
- 7:07 上午
孟德尔是遗传学的奠基人,被称为“现代遗传学之父(father of modern genetics)”,于1865年发现遗传定律。1900年,三位生物学家(德弗里斯、柯灵斯、丘歇马克)推动了孟德尔遗传规律的重新发现。后来人们认识到一些人类单基因遗传病或孟德尔遗传病可以用孟德尔显性和隐性的术语来解释。
在过去十几年中,NGS测序被引入医学诊断中,全外显子测序(WES)为查找致病基因提供了极大的便利。在线遗传学遗传数据库(OMIM)目前列出了6,209种单基因障碍和特征(更新于2022年11月8日),覆盖了70%以上的罕见病(罕见病患病率<1:2,000),影响全球总人口的4~5%。在OMIM目前列出的4,658个常染色体疾病的基因中,约53%(n=2,464)与显性遗传疾病相关,35%(n=1,643)与隐性遗传疾病相关,12%(n=551)与两种遗传模式相关。
2023年2月Nature Reviews Genetics上发表了题为“Mendelian inheritance revisited: dominance and recessiveness in medical genetics”的综述文章[1]。该文阐述了单基因遗传病的遗传变异以及数量效应变异和质量效应变异之间的重要区别,解释了遗传变异在剂量补偿方面表现出的差异和蛋白质变化背后的原理。文章提供了各种临床案例来说明复杂的致病等位基因型与表型之间的关联。最后,文章提出了根据遗传显性和隐性概念来预测遗传变异效应的框架。
根据遗传变异对编码蛋白的功能影响效应进行分类,可将变异可分为数量效应变异(Quantitative variant)和质量效应变异(Qualtitative variant)两大类。前者是变异对所在等位基因编码的蛋白表达量造成影响(表达量增加或减少),但不影响蛋白的三维结构;后者是变异对所在等位基因编码的蛋白结构造成影响。
导致蛋白表达量减少的变异主要有功能丧失变异(Loss of Function variant, LoF)和亚效应变异(Hypomorphic variant, Hyp)。
功能丧失变异(Loss of Function variant, LoF),该变异导致所在等位基因编码的蛋白表达量完全丧失。若LoF为杂合变异,蛋白表达量下降至50%,若LoF为纯合变异,蛋白表达量下降至0%。
单个等位基因LoF变异导致疾病发生的效应,称为单倍剂量不足(Haploinsufficient, HI)。相当于单个野生型等位基因提供的蛋白表达量不足以维持人体的机能。HI对应的遗传方式为常染色体显性遗传(AD)。单个等位基因LoF变异不会导致疾病发生的情况,称为单倍剂量充足(Haplosufficiency, HS)。HS表明细胞内存在补偿机制可以弥补LoF变异造成的蛋白表达量下降。HS对应的遗传方式为常染色体隐性遗传(AR)。
亚效应变异(Hypomorphic variant, Hyp),该变异导致所在等位基因编码的蛋白表达量部分丧失。若Hyp为杂合变异,蛋白表达量介于50%至100%之间。若LoF与Hyp组成复合杂合变异,蛋白表达量介于0%至50%之间。
导致蛋白表达剂量异常增加的突变有三倍剂量敏感(Triplosensitivity)型变异,指的是基因的拷贝数异常增加,过表达的蛋白或基因产物导致生理功能异常。对近100万人的CNV数据进行的分析发现,3635/18641个常染色体蛋白编码基因具有很高的剂量敏感性。
数量效应变异可导致常染色体隐性遗传病,例如苯丙酮尿症(图1)。由数量变异(即杂合子有临床症状)引起的常染色体显性遗传病一般为半显性,在纯合子状态下症状表现更严重。
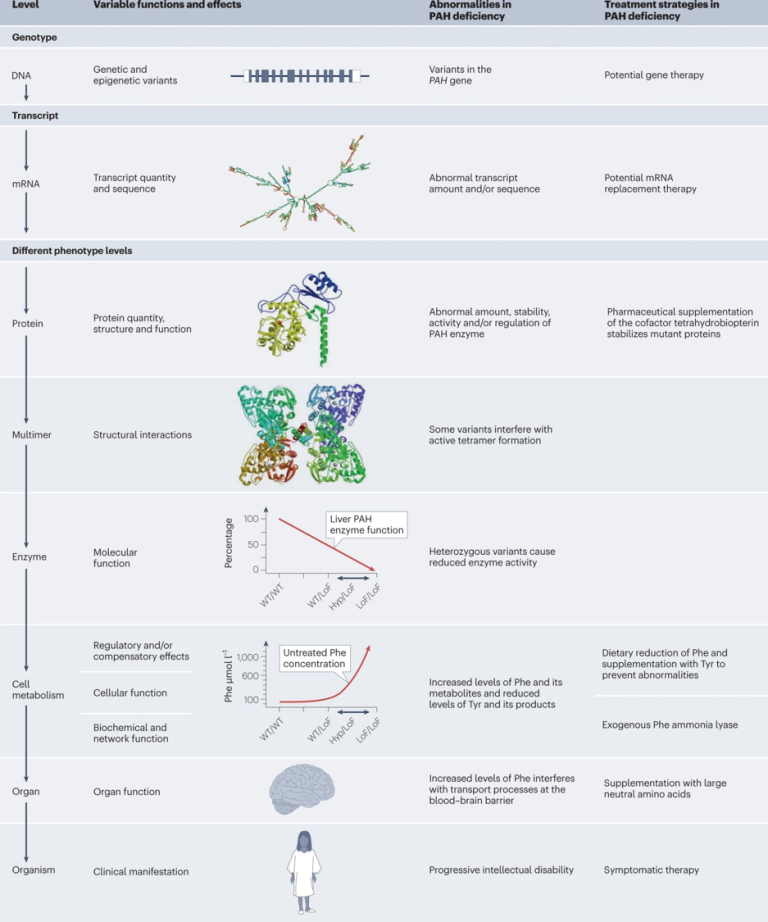
图1 以苯丙酮尿症为例展示从基因变异到临床表型的复杂过程
苯丙酮尿症是一种常染色体隐性疾病,由苯丙氨酸羟化酶(PAH)基因中的致病性变异引起。PAH基因编码的酶催化把苯丙氨酸(Phe)转化为酪氨酸(Tyr)。一个人的整体PAH活性是两个PAH等位基因型综合作用的结果。
在酶的表型水平上,杂合状态(WT/LoF)肝活检中PAH活性降低了50%,代表中间效应。杂合状态(WT/LoF)在代谢方面(血液中的氨基酸浓度:Phe > Tyr,而正常情况下Tyr > Phe)可能会显示出轻微的异常,但在临床表型水平上则完全无症状,符合隐性遗传特征。
PAH基因中2个等位基因功能缺失变异(LoF/LoF)的个体(未接受治疗)血液中Phe浓度极高,会发展成严重的智力残疾。杂合状态下(Hyp/LoF)个体能够维持血液中Phe浓度低于治疗阈值360-600µmol/l,这种情况被称为轻度高苯丙氨酸血症,不需要治疗。苯丙酮尿症通常根据新生儿筛查中的高Phe浓度(即代谢表型)在症状开始前就诊断出,也可以通过基因检测做鉴定。
苯丙酮尿症标准治疗策略是采取低苯丙氨酸饮食,同时补充酪氨酸和其他氨基酸以及维生素和辅因子,这可以保证患儿正常发育并且智力健全。
常染色体基因的数量效应变异基因型通常决定了蛋白功能下降的程度。突变型和野生型(WT)等位基因杂合状态的人表现为显性遗传。功能缺失突变杂合(LoF/WT,50%蛋白活性)状态往往没有症状,表现为隐性遗传(图3)。在半显性遗传中(图3),LoF杂合子通常表现出临床症状,纯合状态(LoF/LoF)受影响更严重。
三倍体表示如果一个常染色体基因有三个拷贝(WT/WT/WT,表现为150%的蛋白功能),例如在三体综合征或微重复中就会出现临床症状。真正的显性疾病在杂合状态和纯合状态中具有相似的临床表现(图2右下),这种现象通常在数量效应变异中观察不到。
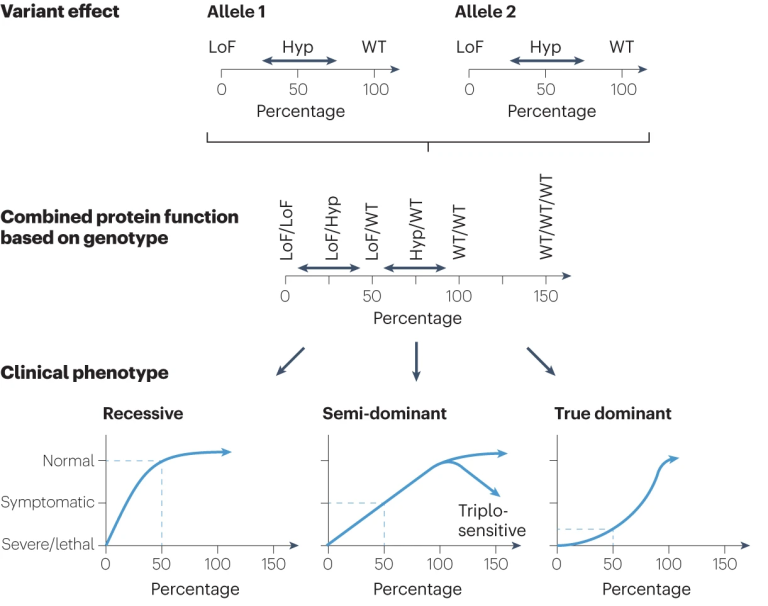
图2 LoF、Hyp基因型对应的蛋白变化情况
假显性遗传是隐性遗传的一种特殊形式,如图3所示,同一家系的多个近亲结合有时会导致连续几代人发生常染色体隐性疾病(家系1)。2号家系中亚效应变异(Hyp)作为多态性变异(Pol)表现为假显性,它与LoF变异杂合致病,但在纯合(Pol/Pol)状态下通常没有症状。
红细胞生成性原卟啉症符合这种遗传模式,该病主要是由FECH基因LoF变异和常见的Hyp剪接变异C.315-48T>C的杂合基因型引起的,在东亚该等位基因频率高达33%[2]。该位点纯合型Hyp剪接变异为良性,受影响的人和带有杂合剪接变体的伴侣的子女的疾病风险为25%。功能性变异用白色文字和蓝色轮廓表示(深蓝色为LoF变异,浅蓝色为Hyp Pol变异),受影响的个体用蓝色圆圈(女性)或正方形(男性;深蓝色为纯合LoF,浅蓝色为杂合Pol/LoF)表示。
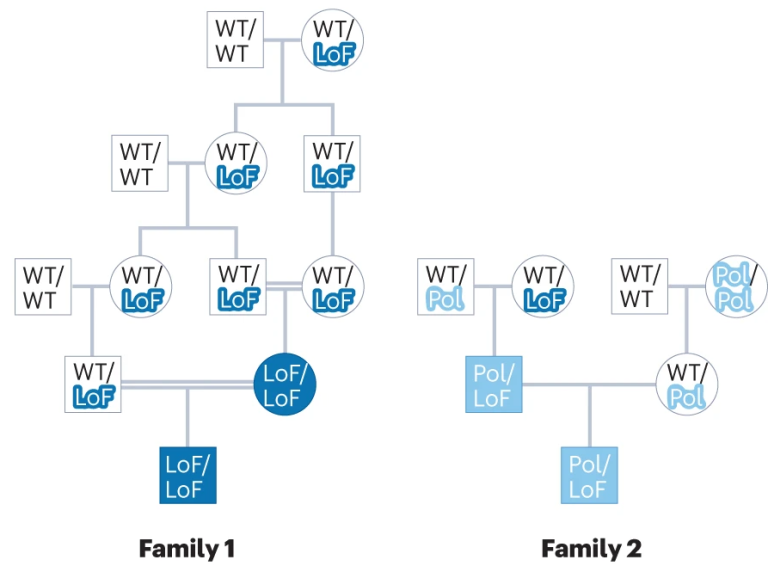
图3 假显性遗传模式家系图
α-地中海贫血是一个双基因遗传的数量性状,人类基因组在染色体16p13.3位置有两个同源的串联α-球蛋白基因,即HBA1和HBA2,它们产生相同的血红蛋白(Hb)α链。因此,五种不同的表型与等位基因的具体组合数量有关。
其中缺失一个或两个基因拷贝无症状或仅为轻度的小细胞性贫血,通常被描述为”α-地中海贫血特征”。缺少三个拷贝会导致HbH病,伴有肝脾肿大,有时会出现输血依赖性贫血,而缺少四个HBA1&2拷贝会导致严重的产前致死性Hb Bart’s hydrops fetalis综合征。
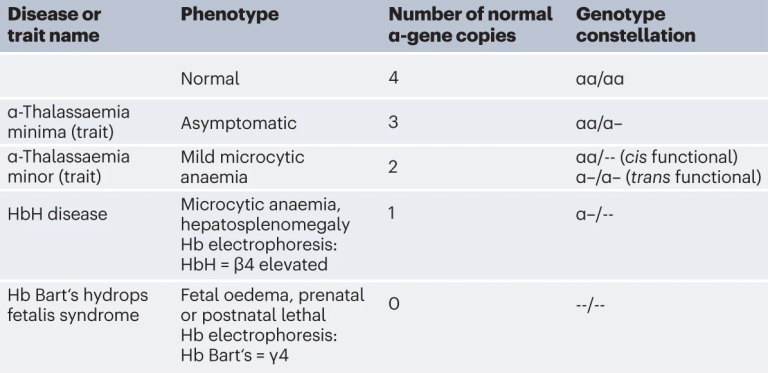
表1 α-地中海贫血的基因型与表现型
许多常见的疾病或定量特征是由许多不同基因的数量效应变异组合引起的[3]。这种情况可以视为多基因性状。只有两个功能性HBA拷贝(αα)的夫妇,其后代的患病风险差异很大,这取决于等位基因型的具体组合形式。图中显示了几种可能等位基因组合及相应的临床症状程度(颜色越深代表严重程度越高)。数量变异效应是许多双基因和多基因性状遗传的基础。
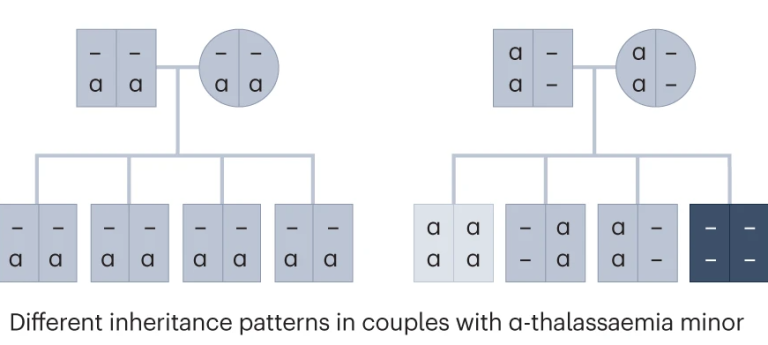
图4 HBA拷贝缺陷夫妇的后代可能基因型及表现型


