


阿尔茨海默病复杂的遗传结构(下)
- boke
- 2025-06-06
- 11:04 上午
阿尔茨海默病(Alzheimer’s disease,AD)是一种复杂的多因素神经退行性疾病,是最常见的认知退化症类型。AD的特征在于淀粉样β肽异常聚集和沉积形成细胞外淀粉样蛋白斑块,以及过度磷酸化tau蛋白形成神经原纤维缠结,继而导致突触和神经元丢失,最终导致进行性认知和功能下降。
AD具有高度遗传性,双胞胎研究的遗传力估计约为70%,APOE和已知的多基因遗传可以解释其中的11%,剩余59%的AD遗传力仍然无法解释[1]。
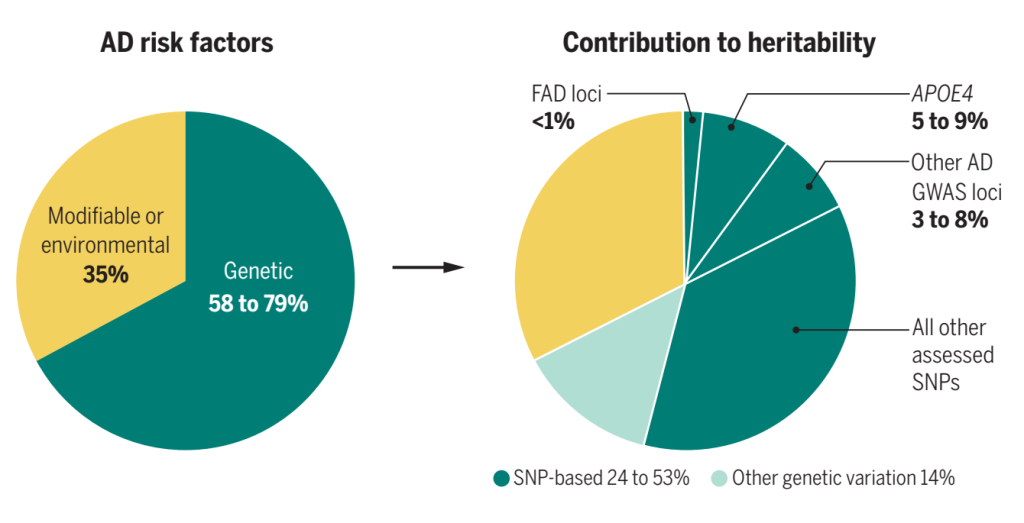
图1. AD风险因素组成[2]。FAD: 家族性AD
不过,近年来,一些有趣的研究正在不断加强我们对AD遗传结构的认知。与传统的APP、PSEN1、PSEN2以及APOE基因变异的“静态”不同,新研究的变异则具有“动态”特征。比如,相对于稳定遗传的 SNV 和 InDel 突变而言,可能在代际间增加的串联重复扩增;或是相对于终生不变的胚系变异而言,随着年龄不断累积的克隆性造血体细胞突变。
“动态突变”(Dynamic Mutation)
动态突变”(dynamic mutation)是一种特殊的遗传变异机制,主要表现为三核苷酸或其他短重复序列拷贝数在代际传递中的不稳定增加或出现组织特异性差异,也称为“重复扩增”。
2024年的一项研究分析了神经退行性疾病(包括AD)与C9orf72基因的重复单元GGGGCC(G4C2)之间的关联[3]。该研究包括27个病例对照研究,纳入了7202名AD患者以及其他神经退行性疾病患者。结果显示,C9orf72重复扩增(>30)导致AD风险升高(OR=4.88),而中度重复扩增(20–30)则与AD无关(OR = 1.16)。研究人员建议,测量C9orf72 G4C2重复扩增可能有助于早期鉴别不同神经退行性疾病。
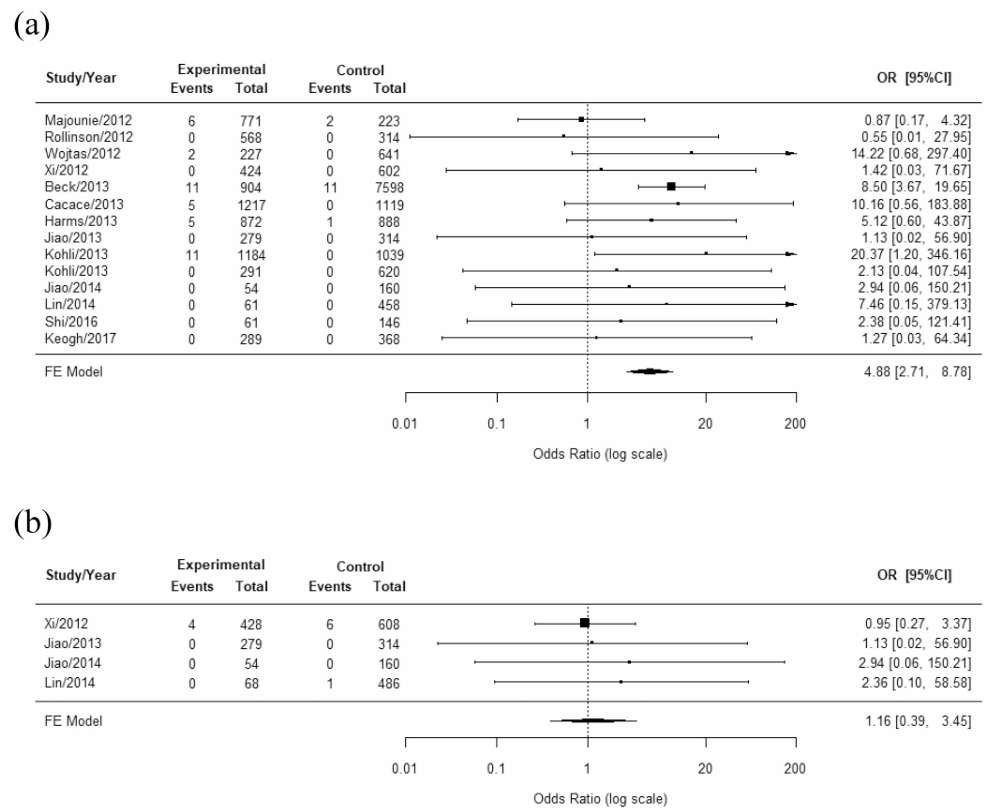
图2. C9orf72重复扩张>30(a)/20-30(b)与AD风险的关联[3]
潜能不明克隆性造血(CHIP)
另一项有趣的研究表明,潜能不明克隆性造血(CHIP)相关突变可以改变AD的进展。
2023年,发表在《自然医学》杂志上的一项研究中,Bouzid及其同事发现了CHIP与预防AD之间的联系[4]。
鉴于全基因组关联研究表明,大脑中的小胶质细胞的功能改变是AD发病机制的主要驱动因素,那么,CHIP是否也与AD风险有关?为了验证这个猜想,研究小组使用血液DNA测序数据,对两个大型队列(Framingham心脏研究和心血管健康研究)进行了全面分析,以研究CHIP与AD的关系。
出乎意料的是,他们发现携带CHIP的人患AD的几率反而较低。同样,这些发现同时在阿尔茨海默病测序项目(ADSP)病例对照队列中得到了验证[OR= 0.64]。
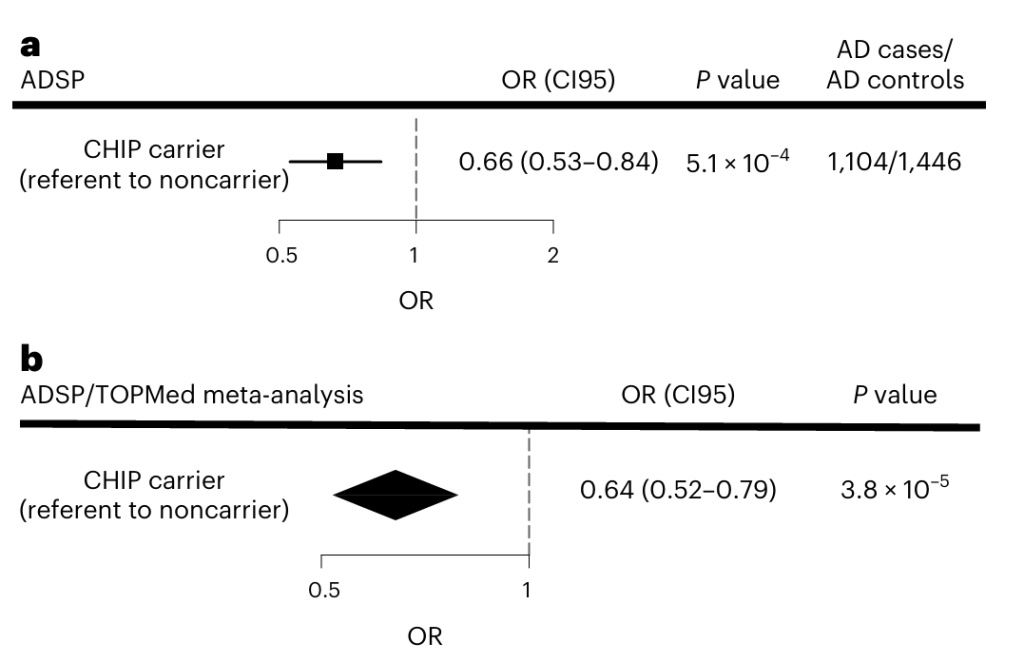
图3. a. ADSP项目中CHIP在APOE ε3ε3 基因型人群中对AD风险的影响;b. ADSP, FHS 和CHS项目中,CHIP对AD风险影响的荟萃分析[4]。
此外,检查阿尔茨海默病相关的病理特征(例如β-淀粉样蛋白斑块和tau神经原纤维缠结)时,他们还发现,在一生中没有出现临床认知退化的人群中,CHIP携带者的确具有较低的神经斑块密度(CERAD分级)和神经原纤维缠结程度(Braak期),CHIP对病理特征的抑制作用与遗传分析一致。
随后,研究小组运用单细胞基因组学技术,探索了小胶质细胞是否可以来源于突变的CHIP克隆。令人惊讶的是,CHIP携带者的大脑中居然含有克隆衍生的小胶质细胞,这与目前普遍认为骨髓细胞不会参与小胶质细胞形成的观点相悖。也就是说,可能是外周髓系细胞进入脑组织,取代了原有的小胶质细胞。
对单细胞测序数据的进一步分析估计,CHIP携带者的突变小胶质细胞比例为30%-90%之间,这表明外周细胞取代内源性大脑驻留的小胶质细胞可能是预防AD的因果机制。
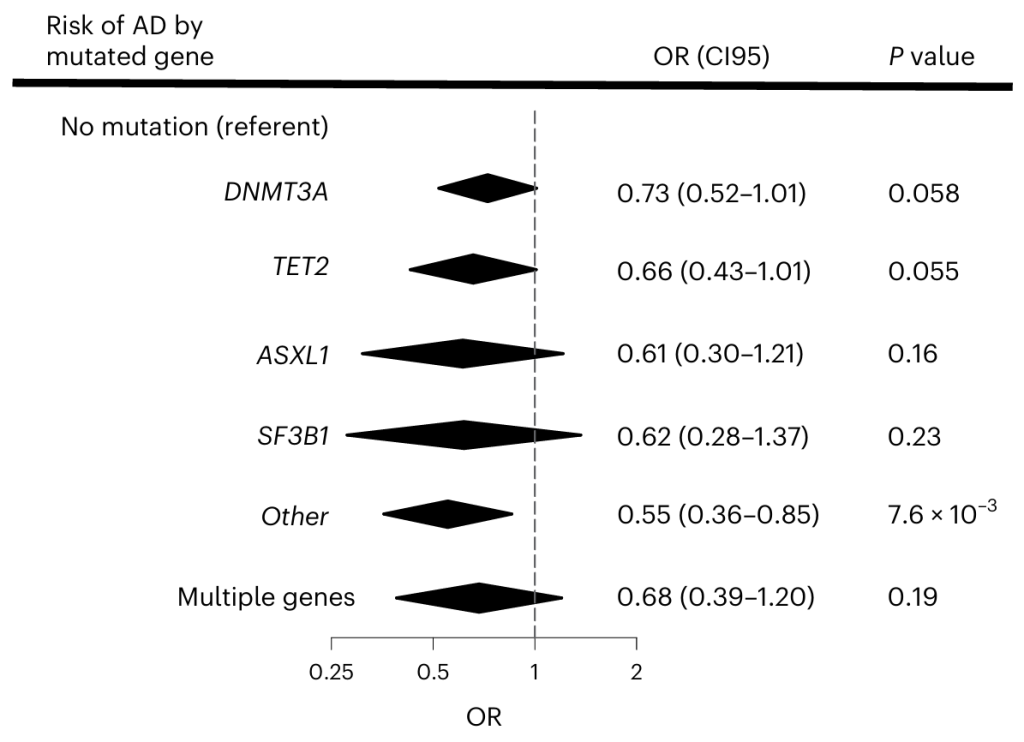
图4. 不同CHIP基因变异对AD风险的影响[4]。
未来,进一步研究携带CHIP的小胶质细胞的机制差异,这可能有助于我们开发治疗AD的新疗法。总之,该项研究揭示了克隆性造血与预防阿尔茨海默病之间意想不到的联系。这些发现为理解阿尔茨海默病背景下的克隆性造血提供了新的视角,并可能为未来开发针对克隆性造血的阿尔茨海默病干预疗法铺平道路。
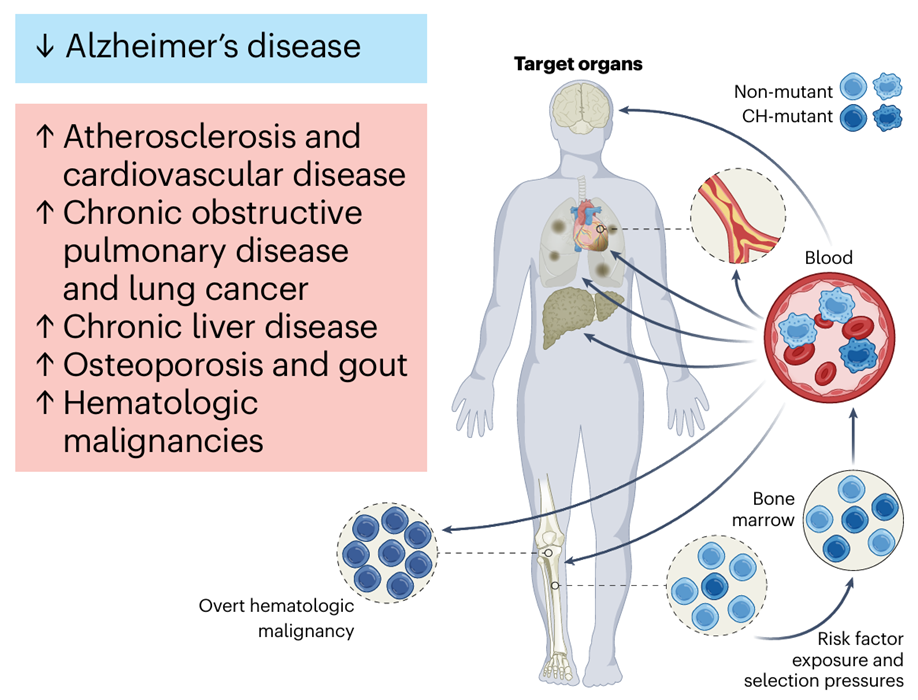
图5. 老年人CH的成因与疾病关联[5]。CH的风险因素包括遗传倾向、高龄、肥胖以及接触烟草、化疗、辐射和感染(右下角)。当这些风险因素出现时,CH突变的造血干细胞(深蓝色细胞)比其野生型对照(非突变;浅蓝色细胞)具有选择性优势。CH突变的造血干细胞能够产生成熟的后代细胞(深蓝色细胞),这些细胞在多个器官中执行免疫功能(箭头所示)。CH突变免疫细胞的活动变化,根据具体情况,可能带来疾病保护(如阿尔茨海默病;左上角蓝色背景框)或增加疾病易感性(如心血管疾病、血液恶性肿瘤等;左上角红色背景框)。
整体来看,AD的遗传学研究有力地证明了一个主要通路的存在,该通路集中于Aβ的产生、聚集和清除,并在早发性和晚发性疾病中发挥作用。遗传学也强烈地暗示了小胶质细胞对淀粉样斑块的反应在AD中的作用。
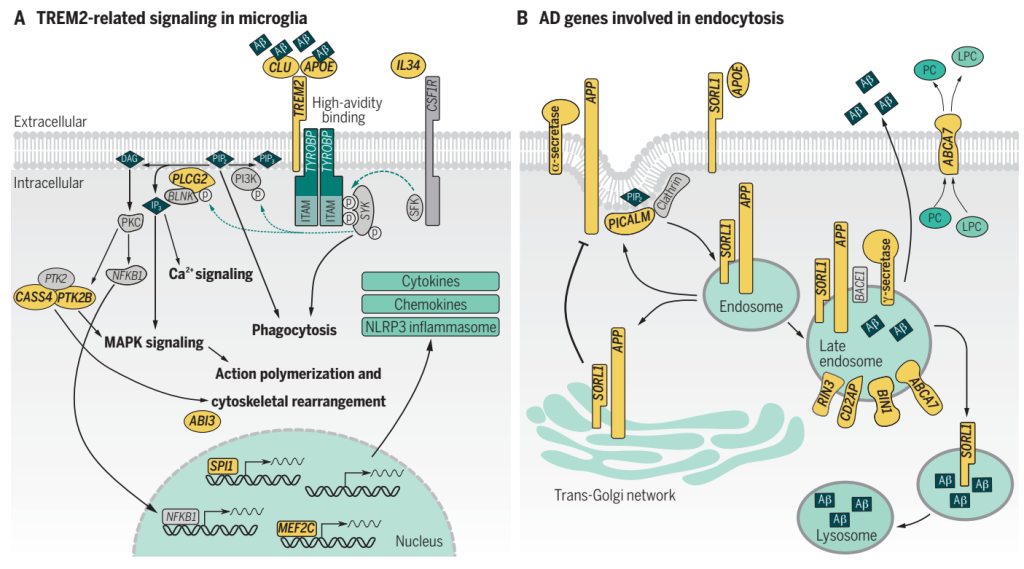
图6. TREM2、SORL1和ABCA7编码的蛋白与其他 AD风险基因(以黄色突出显示)编码的蛋白相互作用,影响小胶质细胞功能和 APP 加工[2]。
研究推测,识别阿尔茨海默症 80% 遗传变异需要超过800万样本量[1]。然而,增加病例数量永远比不上提高质量,需要更深入的临床表型分析和生物标志物研究,以更好地解释遗传变异在特定AD表型方面的作用[2]。
对于一种晚发疾病来说,阿尔茨海默病(AD)风险的遗传成分令人惊讶地巨大。我们在绘制它的遗传图景方面已经取得了巨大进展,现在可以进一步利用过去十年遗传研究中获得的许多线索,构建更复杂的模型,更好地模拟AD的特定机制。寻找治疗像阿尔茨海默病(AD)这样复杂多因素疾病的药物,需要对目标机制有深入的了解。这种思维方式将为药物开发开辟许多机会,并且对患者进行更好的分层将加速从概念到临床的道路。
参考资料
1. Andrews, Shea J. et al. The complex genetic architecture of Alzheimer’s disease: novel insights and future directions. eBioMedicine, Volume 90, 104511
2. Sierksma A, Escott-Price V, De Strooper B. Translating genetic risk of Alzheimer’s disease into mechanistic insight and drug targets. Science. 2020;370(6512):61-66.
3. Jin P, Li Y, Li Y. Meta-analysis of the association between C9orf72 repeats and neurodegeneration diseases. J Neurogenet. 2024;38(1):1-8.
4. Bouzid H, Belk JA, Jan M, et al. Clonal hematopoiesis is associated with protection from Alzheimer’s disease. Nat Med. 2023;29(7):1662-1670.
5. Sánchez Vela P, Trowbridge JJ, Levine RL. Clonal hematopoiesis, aging and Alzheimer’s disease [published correction appears in Nat Med. 2024 Jan;30(1):305.