


阿尔茨海默病复杂的遗传结构(上)
- boke
- 2025-06-03
- 10:10 上午
阿尔茨海默病(Alzheimer’s disease,AD)是一种复杂的多因素神经退行性疾病,是最常见的认知退化症类型。AD的特征在于淀粉样β肽异常聚集和沉积形成细胞外淀粉样蛋白斑块,以及过度磷酸化tau蛋白形成神经原纤维缠结,继而导致突触和神经元丢失,最终导致进行性认知和功能下降。
基因变异在AD的发病中起着重要作用,可以被概念化为位于两个维度上:群体中次要等位基因频率(MAF)和效应大小(图1)。
在这个范围的一端,APP、PSEN1和PSEN2基因中非常罕见的、高致病性突变会导致早发性常染色体显性遗传的阿尔茨海默病(ADAD)。
在范围的另一端,在全基因组关联研究(GWAS)中发现的晚发散发性AD的常见等位基因,它们各自对疾病易感性的影响较小,通常情况下综合起来会增加患病风险。其中,载脂蛋白E (APOE) 被认为是散发型AD风险的单一最大遗传调节因子,影响着平均发病年龄和终生患病风险,携带APOE4 纯合子在65岁左右就会出现AD症状。
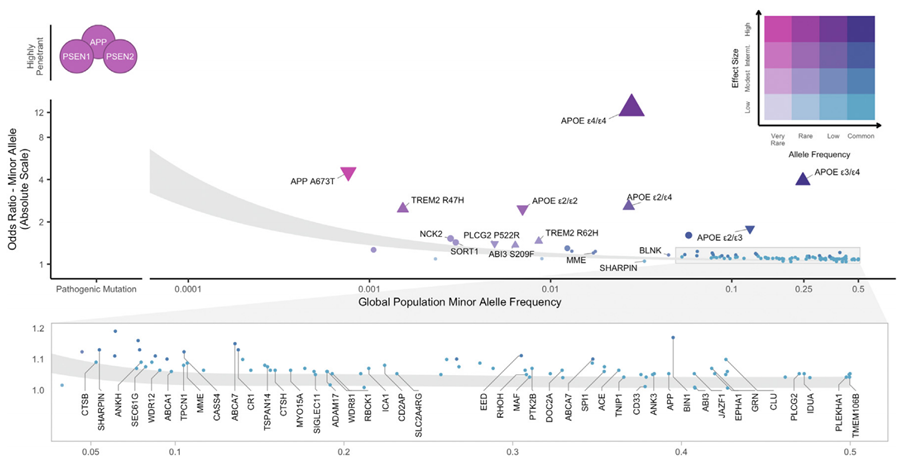
图 1:AD/认知退化的遗传结构,突出显示ADAD突变和81个全基因组水平显著位点[1]。
AD具有高度遗传性,双胞胎研究的遗传力估计约为70%。虽然越来越多的全基因组关联分析 (GWAS) 持续扩展我们对AD/认知退化遗传结构的认识,但直到最近,这些研究才在欧洲血统人群中鉴定了39个疾病易感基因座。
近年来,新的AD/认知退化 GWAS研究显著扩大了样本量和疾病易感基因座的数量。
第一个研究将样本量增加到112.7万人,有效样本量为 33.2 万人,主要包括新的生物库和基于人群的认知退化数据集[2]。
第二个研究则在国际阿尔茨海默病基因组计划 (IGAP) 的早期 GWAS 基础上,增加了临床确诊的 AD 病例/对照数,并纳入了生物库认知退化数据集,总样本量达到78.9万 人,有效样本量为38.2万人[3]。
这两项 GWAS 共同鉴定了90个独立变异,分布在75个AD/认知退化易感基因座中,其中包括42个新的基因座。通路分析显示,易感基因座富集于淀粉样斑块、神经原纤维缠结形成、胆固醇代谢、胞吞/吞噬作用和先天免疫系统相关基因。对新发现基因座的候选致病基因进行了优先排序,确定了62个候选基因。来自已知和新发现基因座的许多候选基因在巨噬细胞中发挥关键作用,并强调了小胶质细胞吞噬清除富含胆固醇的脑组织碎片(吞噬作用)在 AD发病机制中的核心作用,并将其作为潜在治疗靶点。
随着样本量增加, AD相关基因位点数量翻倍(图2)。同时,致病基因筛选方法也取得了显著进展。研究表明,AD相关的基因位点及其候选致病基因参与了不同的生物通路。
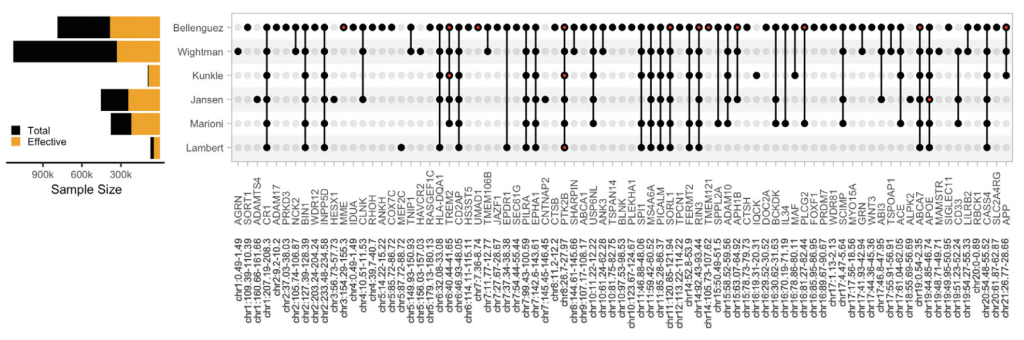
图2. GWAS随着有效样本数量的增加发现了越来越多的相关位点,共同识别了81个基因座上的101个独立变异[1]。全点表示在给定研究中达到全基因组显著性的位点,红点表示包含多个独立相关变异的位点。根据Bellenguez et al.(2022)和Wightman et al.(2022)优先排序的候选因果基因,对位点进行了标记。
阿尔茨海默病基因优先级排序
尽管全基因组关联分析 (GWAS) 加深了我们对 AD 遗传架构的理解,但 GWAS 后功能基因组分析仍然需要筛选影响疾病易感性的基因,并提名候选致病基因进行细胞和动物模型的进一步功能验证。
将早期 AD GWAS 结果与组织和细胞类型特异性表观遗传注释整合,强烈表明髓系细胞(包括脑部巨噬细胞小胶质细胞)在调节疾病易感性方面发挥重要作用。
此外,将 AD 风险变异体与活跃的髓系增强子及其靶基因关联起来,从而筛选出髓系细胞中的候选致病基因(包括 BIN1、RIN3、ZYX、CD2AP、SORT1、CASS4、CCD6、TSPAN14、NCK2)。这些发现也有助于预测AD风险基因表达对疾病的易感性,有助于理解验证实验中基因操作(敲除/敲低或过表达)的结果,并指导开发针对这些AD风险基因的治疗方法。
例如,AD 风险升高与BIN1、ZYX和APPB3表达降低相关,以及与RABEP1和AP4M1表达升高相关。基因调控网络分析也显示,AD风险变异体特异性地影响转录因子 SPI1(PU.1)在小胶质细胞基因调控元件(增强子或启动子)上的结合,SPI1是髓系细胞谱系的主要调控因子。
Wightman 等和 Bellenguez 等进行的基因优先级排序分析确定了另外62个独特的候选致病基因。总之,基因优先级排序结果显示,约51%的AD位点包含参与髓系细胞功能的候选致病基因,进一步表明髓系细胞在AD病因中扮演重要角色。
人类遗传学表明,小胶质细胞吞噬作用和APP代谢是阿尔茨海默病的强烈候选因果途径
人类遗传学将小胶质细胞吞噬凋亡细胞和 APP 代谢确定为阿尔茨海默病强有力的候选致病途径。下一个未解决的问题是 AD 风险变异体如何影响髓系细胞/小胶质细胞的生物学过程,从而调节疾病易感性。
自早期全基因组关联研究以来,常见 AD 风险变异体的通路分析持续显示出富集的基因集,这些基因集参与1) 胆固醇代谢和转运,2) 内吞和吞噬作用,以及 3) 先天免疫系统。最近许多被提名的 AD 风险基因都属于这些生物学通路。
例如,通过整合小胶质细胞数量性状基因座(eQTL)数据,在ECHDC3基因座中的USP6NL被提名为候选 AD风险基因,它参与内溶酶体膜运输。此外,在髓系特异性基因(如TREM2、ABCA7、PLCG2、ABI3)中发现了稀有 AD 风险变异体,这些基因在小胶质细胞和其他髓系细胞吞噬凋亡细胞及其他富含胆固醇的细胞碎片(例如髓鞘碎片和凋亡突触)的先天免疫系统中起着至关重要的作用。
这些发现强烈表明,从全基因组关联研究中出现的三个病因途径并非 AD 的独立致病驱动因素。相反,它们是更高阶生物学过程(吞噬凋亡细胞)的方面,将它们作为髓系吞噬细胞功能紊乱的致病核心组分联系起来(图 3)。
通过整合精细作图分析发现的KCNN4参与小胶质细胞迁移,这对于小胶质细胞识别并吞噬凋亡细胞靶标至关重要。Bellenguez等人的研究提名了其他基因,这些基因支持小胶质细胞吞噬凋亡细胞在AD 中的致病作用。优先基因(例如,TMEM106B、SNX1 和CTSH)的基因本体注释显示出它们与内溶酶体功能的关联。此外,BLNK 已被证明在对吞噬凋亡细胞至关重要的TREM2-PLCG2信号级联中发挥作用。
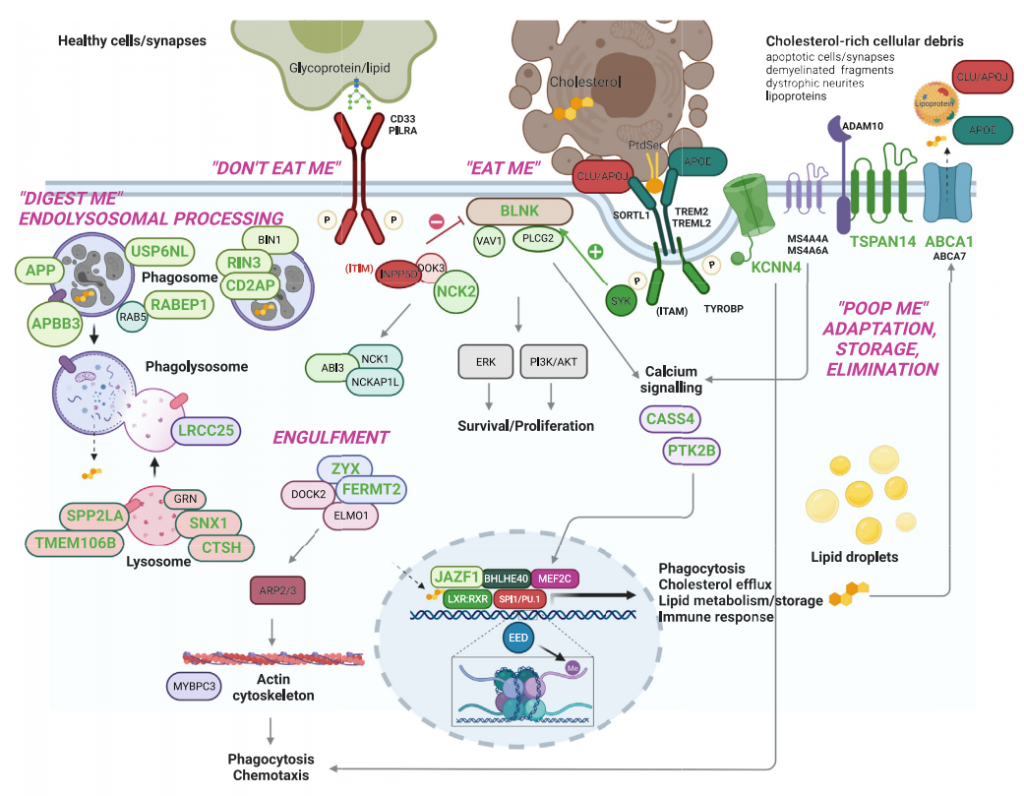
图 3:排名靠前的阿尔茨海默病风险基因及其在小胶质细胞吞噬作用中的作用[1]。与阿尔茨海默病相关的常见和罕见基因变异表明一些基因在巨噬细胞最基本功能之一—吞噬清除死亡细胞及其他细胞碎片(即吞噬作用)中发挥着重要作用。吞噬作用对于维持组织稳态、免疫耐受和炎症消退至关重要。因此,吞噬作用缺陷是多种自身免疫及慢性炎症疾病的病因之一,并可能促成其他疾病的发病机制。吞噬作用是一个多步骤过程,包括:1) 巨噬细胞表面的受体识别死亡细胞,识别“吃我”信号(如磷脂酰丝氨酸,PtdSer)和“不吃我”信号(如唾液酸化糖蛋白/脂质);2) 吞噬摄取死亡细胞进入吞噬体(吞噬);3) 吞噬体与溶酶体融合消化死亡细胞(内溶酶体处理);4) 巨噬细胞对吞噬溶酶体分解产物分子(如胆固醇,其激活 LXR:RXR 核受体以调节参与从巨噬细胞清除胆固醇的基因(如与阿尔茨海默病相关的基因 ABCA1 和 APOE)的表达)进行功能性(如转录、代谢和炎症)适应(清除/代谢)。除了灰色基因外,图中显示参与吞噬作用各步骤的基因,均已通过全基因组关联研究及后续研究与阿尔茨海默病病因相关。绿色基因是通过整合分析筛选出的。因此,人类遗传学证据表明,吞噬作用可能是阿尔茨海默病在巨噬细胞(包括脑部小胶质细胞)中的关键病理机制。
虽然欧洲血统人群的 GWAS 显著提高了我们对AD遗传结构的理解,但基于人群的 GWAS 研究的遗传力估计明显低于双胞胎研究的结果。这种遗传力缺失可能由多种因素共同造成,这表明我们对 AD 遗传结构和遗传风险机制的理解仍有不足。这些知识缺口源于AD研究中一些未充分探索的领域。开展包括不同人群的测序研究,并纳入血液 AD 生物标志物,有望显著提高我们对 AD 遗传结构的认识。
值得注意的是,ATP8B3、JAZF1等基因参与脂质/胆固醇代谢,这是清除凋亡细胞的另一个重要组成部分。与对 AD GWAS 结果的解读一致,最近的研究发现APOE2和APOE4对小胶质细胞的清除凋亡细胞和脂质代谢有相反的影响。此外,最近的研究发现,除了小胶质细胞外,APOE4还在星形胶质细胞和少突胶质细胞中改变了胆固醇的平衡和运输。
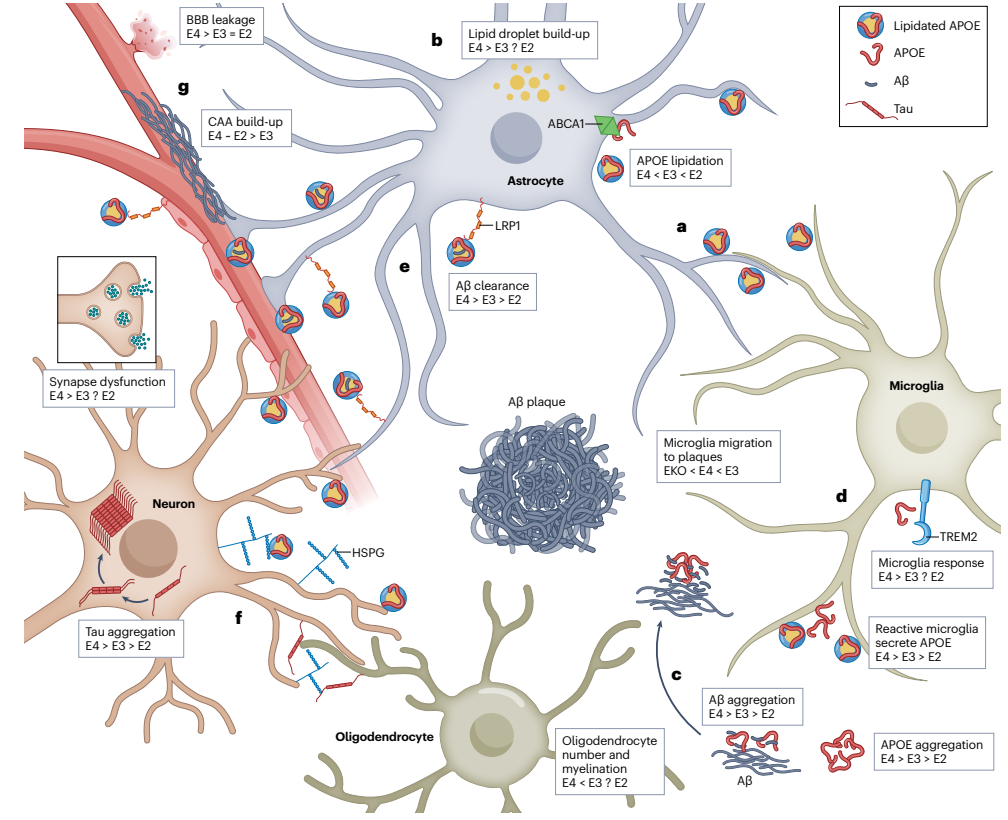
图4. APOE 在阿尔茨海默病病理生理中的多方面作用[4]。载脂蛋白 E (APOE) 主要由健康脑组织中的星形胶质细胞产生,但在阿尔茨海默病患者脑组织中的反应性小胶质细胞中也有产生。
a, APOE 主要由位于星形胶质细胞质膜中的脂质转运蛋白 ABCA1 脂化。b, APOE4 促进星形胶质细胞胞浆中脂滴的积累,这表明星形胶质细胞的脂质转运和能量代谢受损。 c, APOE 与淀粉样蛋白 β (Aβ) 肽相互作用,APOE4 促进其寡聚化、纤维化,并在 Aβ 斑块中成核,并加剧脑淀粉样血管病 (CAA) 的发生。 d, APOE 与小胶质细胞质膜中的 TREM2 相互作用,影响小胶质细胞对 Aβ 和 tau 的反应,例如,影响小胶质细胞向斑块迁移,以等位基因依赖性方式进行。 e, APOE 被星形胶质细胞、神经元和内皮细胞摄取,这也被认为与神经元摄取 tau 相关,均通过 LRP1 介导的内吞作用。 f, 细胞外 tau 和 APOE 可能竞争性地结合神经元表面上的硫酸乙酰肝素蛋白聚糖 (HSPGs),这可能对 tau 的传播有潜在影响。 g, 除了脑淀粉样血管病 (CAA) 外,APOE4 还促进血脑屏障 (BBB) 破坏,导致血浆蛋白和微出血。
APOE4纯合子AD
2024年,在Nature Medicine上,Fortea等人提供了全面的证据[5],支持将APOE4纯合子阿尔茨海默病重新定义为一种新的遗传性阿尔茨海默病,类似于早发性常染色体显性阿尔茨海默病(ADAD)和唐氏综合征相关阿尔茨海默病(DSAD)(图5)。
APOE4纯合子约占总人口的2%,并且占所有阿尔茨海默病病例的15%。因此,APOE4纯合子阿尔茨海默病很可能成为全球最常见的孟德尔疾病之一。这一事实不仅可以提高公众对阿尔茨海默病的认识,还可以促使诊断、管理和护理策略发生根本性转变。这对APOE4纯合子个体来说也是至关重要的信息,并可能促使开发教育和咨询项目以支持这些个体。将APOE4纯合子阿尔茨海默病重新定义为一种遗传形式的阿尔茨海默病,将对阿尔茨海默病的诊断、研究和治疗发展产生重大影响。
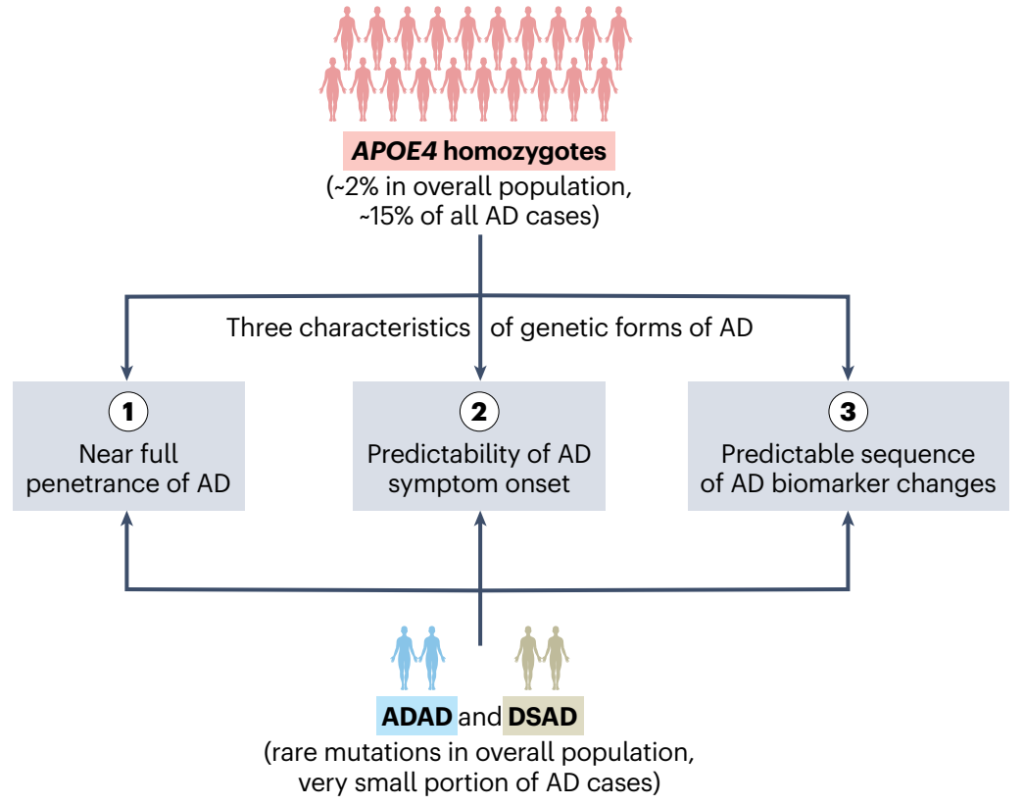
图5. 重新定义阿尔茨海默病的遗传形式[5]。APOE4 纯合子约占总人口的 2% 以及所有阿尔茨海默病病例的15%。应将APOE4纯合子归类为阿尔茨海默病的一种遗传形式,类似于ADAD,其由 APP、PSEN1 或 PSEN2 基因的基因突变造成,导致淀粉样β肽产生变化,并形成淀粉样斑块;以及DSAD,由 APP 基因三倍体化引起。
靶向载脂蛋白E(APOE)的治疗方法
鉴于APOE4等位基因在欧洲和东亚人群中频率较高,且对AD风险影响显著,以及其对大脑功能的多种负面影响,降低APOE4水平是预防或延缓AD进展的合理策略。
图6展示了展示了在小鼠模型中测试的主要针对APOE的治疗策略。
3a:使用小分子模拟Aβ-APOE结合域来阻断与APOE的相互作用;
3b:通过腺相关病毒(AAV)载体递送APOE2来提高APOE2水平;
3c:使用抗APOE抗体降低APOE4水平;
3d:使用反义寡核苷酸(ASOs)降低APOE4水平;
3e:通过小分子肽模拟物上调ABCA1以增加APOE的脂化水平;
3f:使用小分子纠正APOE4的构象,使其更接近APOE2。
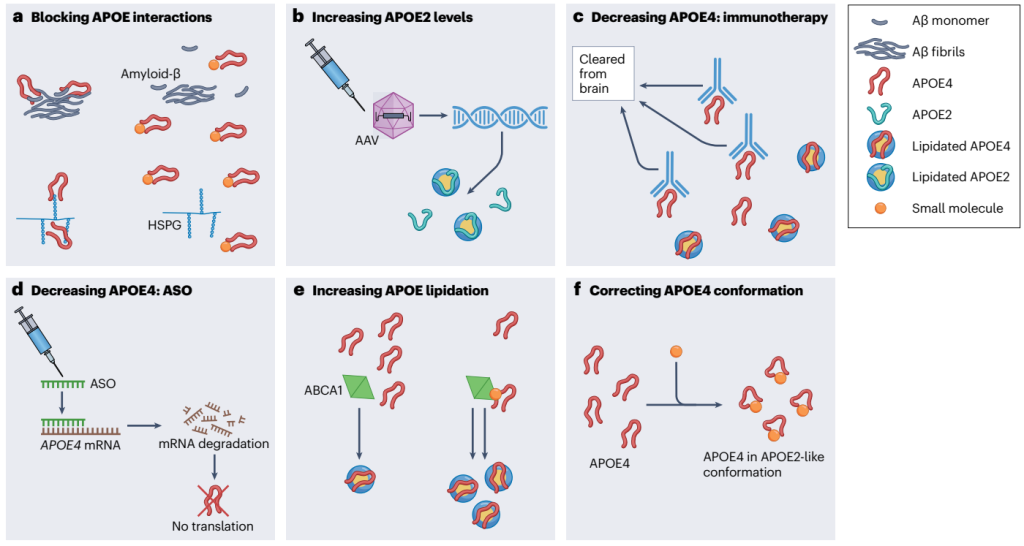
图6. 针对APOE的治疗策略[4]
总之,跨多个人群的AD全基因组关联研究(GWAS)已鉴定出超过80个基因座,但其中大部分位于欧洲血统队列中。这些发现极大地提升了我们对AD遗传基础的理解,并揭示了小胶质细胞吞噬作用和APP代谢在AD发病机制中的作用。重新定义的新的遗传性AD(APOE4纯合子)将对阿尔茨海默病的诊疗发展产生重大影响。
然而,仍有很大一部分AD的遗传机制有待发现。进行大规模WES/WGS数据分析有助于识别与AD相关的稀有变异。增加AD GWAS中的种族和民族多样性将提高遗传发现和多基因风险评分在不同人群中的适用性,帮助阐明遗传风险机制,并促进特定人群的遗传效应的发现。收集血液生物标志物有助于提高诊断准确性,而血液生物标志物GWAS将推动发现影响特定AD生物机制的遗传变异。
虽然我们专注于检查遗传因素对AD的贡献,但环境和生活方式因素估计占表型差异的29% ,然而,它们在AD遗传易感性方面的影响在很大程度上仍然未知。大规模整合研究,结合遗传学、多组学、生活方式风险因素和健康社会决定因素,对于对AD病理机制的理解和公平的治疗发现至关重要。
参考资料
1. Andrews, Shea J. et al. The complex genetic architecture of Alzheimer’s disease: novel insights and future directions. eBioMedicine, Volume 90, 104511
2. Wightman DP, Jansen IE, Savage JE, et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat Genet. 2021;53:1276–1282
3. Bellenguez C, Küçükali F, Jansen IE, et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat Genet. 2022;54:412–436.
4. Jackson, R.J., Hyman, B.T. & Serrano-Pozo, A. Multifaceted roles of APOE in Alzheimer disease. Nat Rev Neurol 20, 457–474 (2024).
5. Xu, Q., Liang, Z. & Huang, Y. APOE4 homozygosity is a new genetic form of Alzheimer’s disease. Nat Med 30, 1241–1242 (2024).