


综述| 非翻译区域变异在孟德尔疾病中的作用(上)
- boke
- 2025-08-12
- 2:20 下午
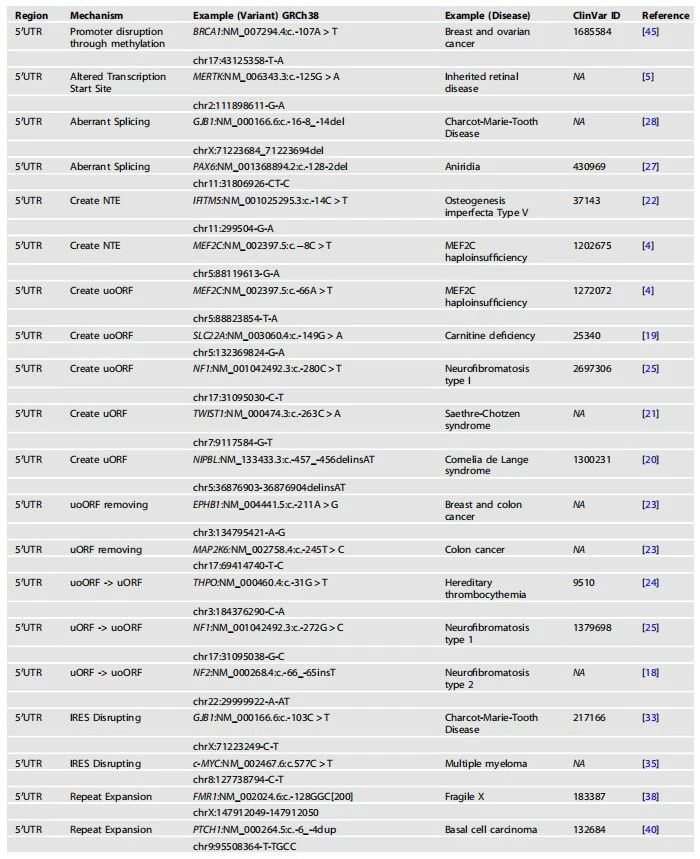
非翻译区(UTRs)位于基因的蛋白质编码序列两侧。 5’UTR和3′UTR序列通过线性结构和结构元件介导转录后调控,控制RNA的稳定性、细胞定位和蛋白质翻译速率。5’和3’UTR内的变异已被证明可以通过多种不同的机制导致疾病。
然而,为了使这些变异能够常规地注释和解释在临床遗传检测中,我们需要更好地了解这些区域及其中的致病变异谱。在本综述中,我们系统地评估了先前在UTRs中识别出的孟德尔致病变异,并列出了其潜在机制。随着基因组测序的广泛应用和越来越多地应用于诊断环境,本综述将为考虑和解释UTR变异提供宝贵资源。
引言
确定疾病的遗传基础对于患者的临床管理至关重要,可能提供治疗靶点,并使患者及其家庭了解生育决策。迄今为止,临床遗传学主要关注直接编码蛋白质的基因组区域的变异,严重罕见疾病的遗传诊断率为 ~30-50%。尽管已知非编码区域的变异也可能导致疾病,但这是一个相对研究较少的领域,只有最近才发表了支持非编码区域变异常规临床分类的建议。
此综述聚焦于未翻译区(UTRs)内的变异及其在孟德尔疾病中的作用。虽然UTRs长期以来被称为“非翻译”,但我们现在知道,和UTR内的调控元件都经历着活跃的翻译。因此,这些区域可能更恰当地被称为“前导”序列(5’UTR)和“尾部”序列(3’UTR)。然而,在这里我们使用更熟悉的UTR术语。
UTRs是直接位于基因蛋白质编码序列(CDS)两侧的非编码区,它们被转录成mRNA但不翻译成标准蛋白质。它们在基因调控中发挥着重要作用,并且在基因间高度可变。
理解UTR内的功能调控元件对于理解其在翻译和功能中的作用以及扰动如何导致疾病至关重要。通过增加对这些区域的知识,我们可以开始将这些区域的分析纳入临床遗传学,并提高诊断率。因此,在这里,我们系统地综述了和UTR中的调控特征,这些特征在受到破坏时已被证明或假设会导致孟德尔疾病。我们排除了先前已审查的更常见、更复杂的疾病。
UTRS是重要的调控元件
蛋白质生产是细胞中精细调节的过程;过多或过少都可能扰乱细胞过程并导致疾病。5’和3’非翻译区(UTR)共同在调节蛋白质生产中发挥关键作用,通过控制转录稳定性以及蛋白质合成的速率和位置。
5’UTR
5’UTRs的主要功能在于翻译调控。mRNA翻译成蛋白质的过程通常从mRNA的5’帽子上招募核糖体开始,核糖体沿着mRNA从5’到3’方向扫描(即通过转录前导序列),并在起始密码子处启动翻译,通常是一个AUG。这个过程通过上游开放阅读框(uORFs)和影响蛋白质产量多少及速度的结构特征等关键序列元件进行调控。
5’UTR的长度,以及其中各种翻译调控元件的数量和类型,在基因之间差异很大,为每个基因提供正常蛋白质生产所需的正确元素组合。尽管5’UTRs平均来说非常短(平均约200bp),但在某些基因中,它们的长度甚至超过了编码序列(CDS)。通常,在需要仔细控制剂量重要的基因中,5’UTRs更长且更复杂。
选择性剪接和不同的转录起始位点(TSS)使用会导致包含不同翻译调控元件组合的多样化的 UTR 异构体,从而在不同的组织和发育阶段实现不同水平的蛋白质翻译。
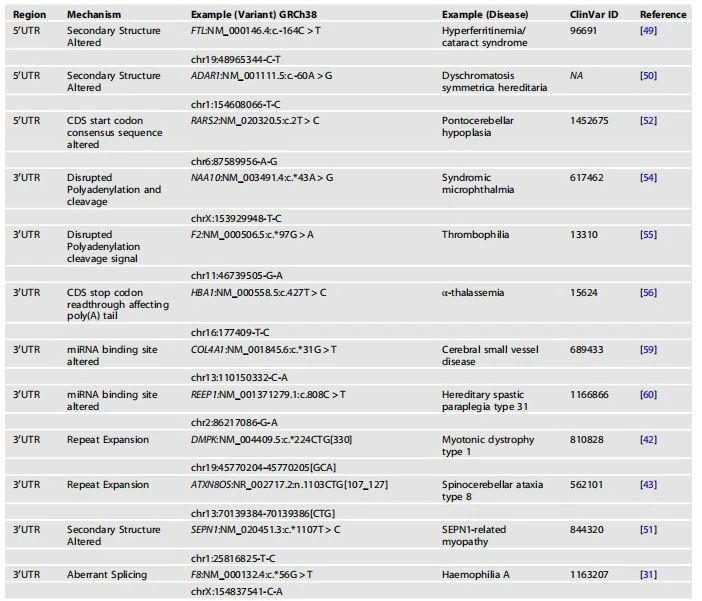
3’UTR
3’UTRs 的主要作用在于调节 mRNA 分子的稳定性、其降解速率以及其在细胞内的位置。尽管,与 5’UTRs类似,它们也在翻译调节中发挥作用。但3’UTRs的这种调节大多是通过与额外因子的相互作用来介导的,尤其是与3’UTRs内的基序和结构元件结合的microRNA(miRNA)和 RNA-Binding Proteins(RBPs),以介导其效应。
虽然 miRNA 通常具有抑制作用,但 RBPs 具有多种不同的调节作用。与5’UTRs一样,不同基因以及同一基因的不同转录异构体之间,调节元件的精确组合差异很大。可变多聚腺苷酸化(Alternative Polyadenylation,APA)可以产生具有不同长度 3’UTRs 的转录异构体,这些转录异构体含有不同数量的调节性 miRNA 和 RBPs 的结合位点。
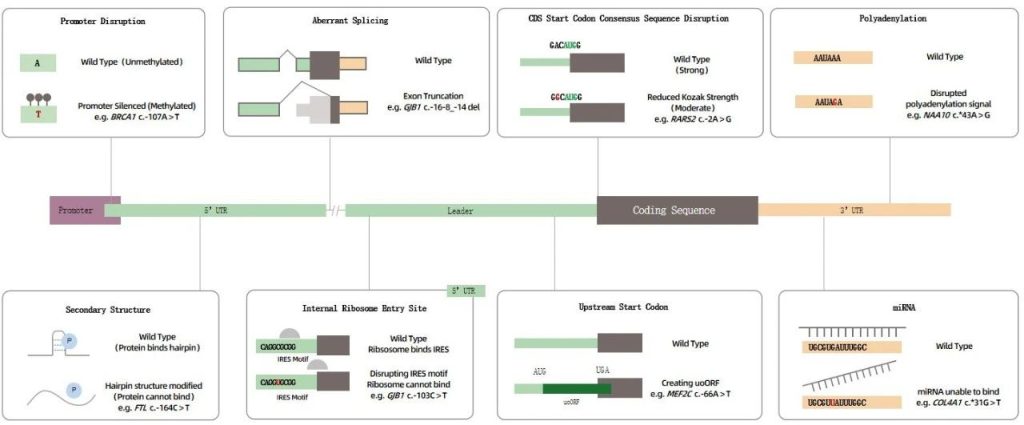
图1 UTR调控元件及导致疾病的变异示例。基因和蛋白质表达受到严格控制,当这些调控受到破坏时,可能导致疾病。
考虑到UTRs中调节元件的关键作用,通过基因变异扰动一个或多个上述元件可能会对蛋白质产生重大影响,并导致严重疾病(图1)。以下各节回顾了迄今为止已发现的多种潜在机制。表1列出了用作例子的所有变异。
表1. 孟德尔疾病中UTR变异的例子。
变异形成了新的起始密码子导致正常CDS的翻译减少
上游AUG(uAUG)三联体通常存在于5’UTR中。uAUGs或其他近缘密码子(最常见为CUG)可能被扫描核糖体识别为起始密码子,从而启动翻译。
从上游起始密码子翻译可能产生多种效果(图2A)。在CDS开始之前,如果起始密码子有一个阅读框内的终止密码子时,即编码uORFs(例如位于5’UTR内)。uORF的翻译可能随后是核糖体从mRNA上解离(因此降低CDS翻译)或继续扫描,在下游CDS重新启动翻译。如果没有框内终止密码子,这个上游起始的翻译将与CDS重叠。如果这个上游的起始与CDS起始密码子同阅读框,从它开始的翻译将导致N端延伸的蛋白质(N-terminal extension,NTE)。或者,如果上游起始与CDS不同阅读框,上游重叠的ORF(uoORF)将会在在CDS内终止或者跨越CDS终止密码子后终止。
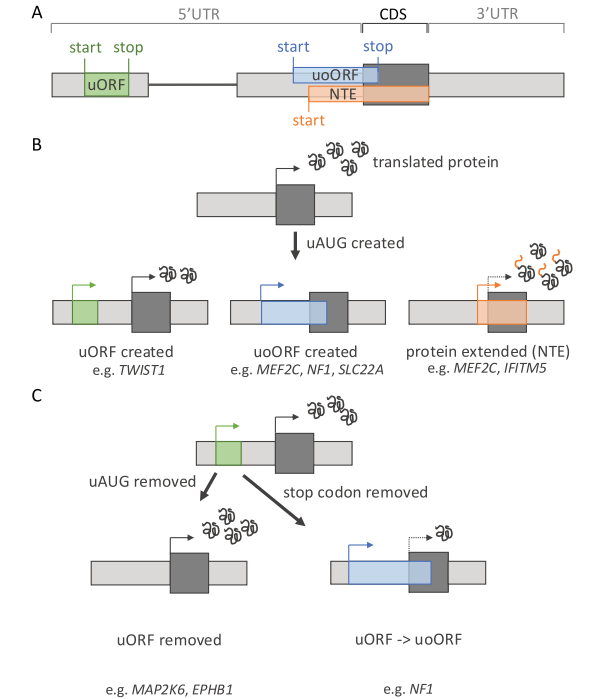
图2 “创建”和干扰上游开放阅读框(uORF)的致病机制。
上游起始密码子的翻译通常被认为会抑制CDS翻译,因为已经证明uORFs的活性翻译可以减少下游CDS翻译高达80%。uAUG通过影响核糖体对起始密码子的识别而启动翻译的前景取决于局部序列环境,即Kozak序列。如果AUG没有Kozak环绕,那么这个AUG很容易被核糖体跳过。
大约43%的基因在其5’UTR中有一个或多个uAUG,这些uAUG在5’UTR中的保守性比任何其他三联体都要高得多。新uAUG的产生已被证明受到强烈的负选择。特别是,创建uoORFs或NTEs的变异,总的来说,与错义变异一样有害。
变异导致产生框外uORFs已在多种疾病中发现。这包括导致肉碱缺乏的隐性基因SLC22A(SLC22A:NM_003060.4:c-149G>A),其中多个患者为复合杂合子,其中一个uAUG-产生变异位于5’UTR,另一个CDS变异均预测会降低蛋白质水平。其他例子包括MEF2C(NM_002397.5:c-66A>T)和NF1(NM_001042492.3:c-280C>T),这些uoORF的变异会分别导致严重的发育障碍和神经纤维瘤病I型。
如果形成了有利于上游起始密码子的有利序列环境而使得uoORF高表达,这可能导致CDS翻译完全丧失。相反,如果由于称为“扫描遗漏”(‘leaky scanning’,没有Kozak序列环绕的AUG,这个AUG很容易被核糖体跳过的现象)的过程而导致上游起始密码子的翻译起始不完全,该变异将只会导致蛋白质水平部分降低。此类表型减弱的变异是否具有足够大的影响以引起疾病,取决于每个基因的剂量敏感性水平,从而使得变异解释复杂化。
“uORF-创建”变异也被报道会导致疾病,再次作用于降低CDS翻译。例如,NIPRL中的变异导致Cornelia de Lange综合征(NM_133433.3:c- 457_ – 456delinsAT),以及TWIST1(NM_000474.3:c- 263C > A)导致Saethre- Chotzen综合征。
然而,“uORF-创建”变异的解释更加困难,因为引入新uORF对CDS翻译的影响难以预测。不仅uAUG上下游影响uORF翻译的强度,而且新形成的uORF的长度以及uORF末端与CDS起始密码子的距离都会影响核糖体在CDS起始密码子下游重新启动翻译的可能性。值得注意的是,所有uAUG-产生变异的解释还因5’ UTR的复杂组成而进一步复杂化。例如,如果在已经高度翻译的uORF中形成uAUG,它可能不会被扫描核糖体“看到”作为潜在的起始位点。或者,形成uORF阻止了具有更强抑制作用的现有uORF的翻译,可能导致CDS翻译的上调。
虽然uORF和上游重叠ORF(uoORF)产生的变异会影响翻译调控,但在CDS阅读框内创建uAUG的变异,如果导致NTE,则可能更倾向于破坏蛋白质功能。
例如,一个变异(NM_001025295.3:c.- 14C > T),通过增加5个氨基酸延长了IFITM5的N端,在患有成骨不全V型的个体中反复出现。在这种情况下,额外氨基酸的添加使蛋白质失去功能。
同样,两个不同的变异分别向MEF2C蛋白的起始处添加了3个(NM_002397.5:c.- 8C > T)和9个(NM_002397.5:c.- 26C > T)氨基酸,导致MEF2C单倍体不足和严重的发育障碍。MEF2C无法容忍N端的氨基酸添加,因为这可能会破坏该转录因子与DNA的结合,从而废除其功能。
变异可能影响现有的uORFs或uoORFs,从而扰乱翻译调控
以上“uORF-创建”变异的例子均预测会降低CDS翻译。相反,移除现有的上游起始密码子更有可能增加蛋白质水平并产生获得性功能效应。在癌症样本中发现的EPHB1(乳腺和结肠)(NM_004441.5:c.- 211A > G)和MAP2K6(结肠)(NM_002758.4:c.- 245T > G)上游起始密码子移除变异(分别移除uoORF和uORF)与增强翻译相关,表明uAUG丢失介导的下游主要蛋白编码序列翻译增加可能有助于致癌。
变异也可能改变uORF或uoORF的抑制效应。例如,THPO(NM_000460.4:c.-31G > T)5’UTR中的变异通过在uoORF序列中创建一个终止密码子,将uoORF转变为uORF。原本的uoORF是被强烈翻译,用于保持THPO的低表达。THPO编码血小板生成素,对于控制血小板生成的通路正常功能是必需的。将uoORF转变为uORF(可能重新启动)会增加蛋白质水平,导致遗传性血小板增多症。
相反,将高度翻译的uORF转变为uoORF可以增加其抑制作用。这可以通过两种不同的机制实现。第一种机制的一个例子在NF1中可以看到,其中观察到删除了原生uORF的终止密码子(例如,NM_001042492.3:c.- 272G > C)的变异体,将其转化为uoORF(因为CDS之前没有其他in-frame终止密码子作为替代),在神经纤维瘤病I型患者中存在。第二种机制在NF2中观察到,其5’ UTR包含一个具有先前翻译证据和强预测Kozak共识的原生uORF。一个单碱基插入(NM_000268.4:c.- 66_65insT)改变了uORF的阅读框,使其绕过下游的终止密码子并创建一个out-of-frame的uoORF。预测这种uoORF的翻译将降低NF2的翻译,这与导致神经纤维瘤病II型的单倍体不足一致。类似于创建uAUG的变异体,解释改变现有uORF的变异体可能很复杂。uORF和uoORF以及不同长度的uORF所传递的翻译抑制差异难以预测。
变异可以通过改变UTR剪接引起疾病
UTR区内的变异通过多种机制引发疾病(图3)。大约38%的5’UTR包含内含子,内含子数量可达11个。已知5’UTR的剪接变异会影响mRNA的稳定性和翻译。
数个影响PAX6的5’UTR内剪接的变异被认为与无虹膜症有关。这些变异(例如NM_001368894.2:c.- 128- 2del)导致5’UTR的2和3外显子附近的剪接跳跃或错误。疾病的假设机制是通过uORF失调(图3A);存在一个跨越这些外显子的uORF,据报道,这些变异改变了uORF的阅读框,类似于上述的NF2例子,将uORF转变为更具抑制性的uoORF,导致蛋白功能丧失和疾病。
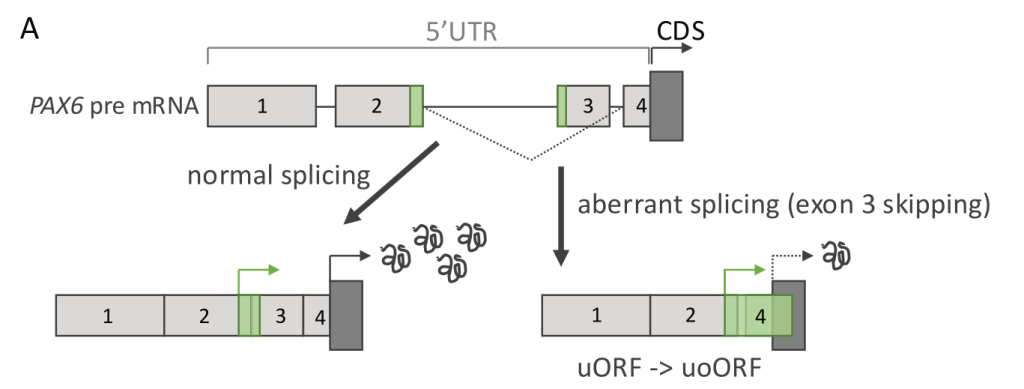
图3A. PAX6中的UTR变异是无虹膜症的常见原因。这些变异被认为造成了第三外显子跳跃,因为其含有上游开放阅读框(uORF)的终止密码子,这就将其转换为上游重叠开放阅读框(uoORF),从而降低PAX6编码序列(CDS)的翻译。
改变5’UTR剪接也可能影响CDS序列。例如,破坏5’UTR中终止剪接位点的变异会导致包含CDS起始密码子的外显子被跳过或截短。一个例子是GJB1(NM_000166.6:c.-16-8_-14del),其中这种机制导致后续外显子被大量删除(278 bp),包括CDS的262 bp(31%;图3B)。据报道该变异会导致Charcot-Marie-Tooth病。
虽然5’UTR中剪接很常见,但只有大约6%的3’UTR包含内含子。大多数内含子都非常靠近CDS终止密码子,因为CDS下游50/55 bp以上的内含子会导致转录本通过无义介导降解(NMD)途径降解。3’UTR中创建新内含子的变异可以触发NMD,并导致蛋白质表达降低。例如,F8(NM_000132.4:c.*56G > T)中的变异创建了一个新的供体剪接位点,导致3’UTR中159 bp的缺失,已被证明会降低表达,因此会导致轻度血友病A。
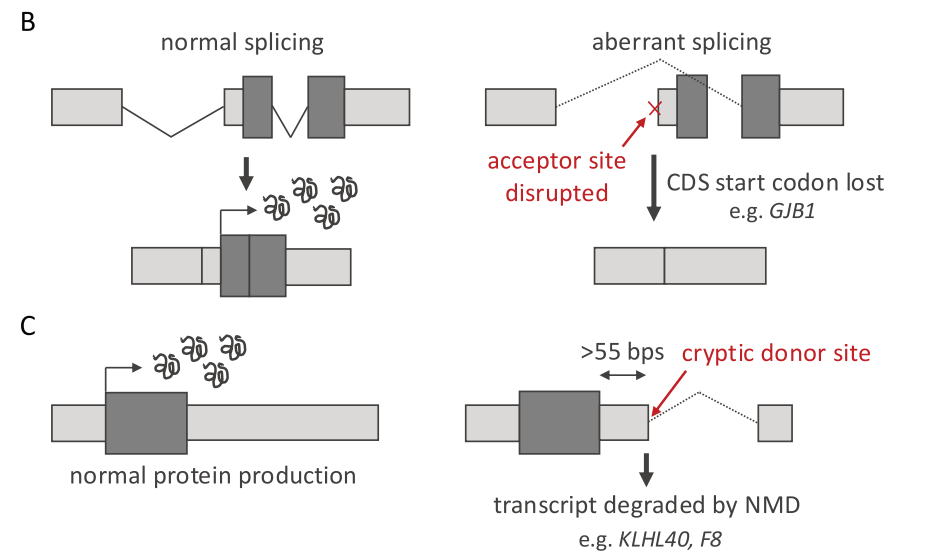
图3B.破坏UTR中的剪接受体位点的使得CDS的起始密码子被移除;图3C. UTR变异在CDS末端下游55bp以外创建隐匿供体剪接位点,预计会导致通过无义介导的降解(NMD)导致转录本降解。
内部核糖体进入位点(IRES)的变异可能影响核糖体招募
一部分mRNA异构体可以通过帽依赖性方式通过内部核糖体进入位点(IRESs)启动翻译。这些是位于5’UTR内的特化序列,可以直接招募40s核糖体亚基,从5’UTR内部独立于5’帽启动扫描。值得注意的是,IRES基序在不同基因之间存在差异,因此难以预测,限制了我们对这些元件变异的注释和解释能力。此外,尽管有许多生物体中有IRES的记录实例,但支持它们在人类中广泛存在的证据有限。
可能正因于此,关于IRES在疾病中的作用在已发表的文献中很少提及。以与Charcot-Marie-Tooth病相关的GJB1中的一个变异(NM_000166.6c.- 103C>T)为例,GJB1的原生IRES对于神经细胞中connexin-32 mRNA的翻译至关重要,该变异据报道消除了IRES的功能,导致无翻译,这是通过使用双顺反子报告基因进行的深入的体内分析发现的。然而,这种IRES的存在因最近的研究实验而受到质疑。另一个例子是在原癌基因c-myc中,在患有多发性骨髓瘤的患者中,报道的IRES中一个 C>T的替换(NM_002467.6c.577 C>T)导致IRES活性升高和c-myc蛋白的产生增加。其潜在机制尚不完全清楚,但实验数据表明,c-myc IRES转座作用因子(Y-盒结合蛋白1(YB-1)和多聚嘧啶轨道结合蛋白1(PTB-1))与突变型c-myc IRES的结合更强。值得注意的是,新的转录注释将这种变异分类为CDS中的错义变异。
重复的UTR扩展可能会扰乱调控并导致毒性肽的产生
重复扩增(也称为微卫星或简单序列重复)是一类独特的变异。核苷酸序列、基因内的位置、重复长度的范围和临床结果在不同重复之间有所差异。当位于非编码区时,重复扩增可能是致病性的,导致所谓的非编码重复扩增疾病,其中UTRs内有许多例子。
致病机制多种多样,但主要可能的机制如下:重复序列可以形成影响转录、翻译和与各种RNA结合蛋白(RBPs)结合的分子内结构;富含GC的重复序列也容易发生高甲基化,从而导致基因沉默;更少见的是,通过与重复相关的非ATG(RAN)翻译,重复RNA本身可能被非传统地翻译成有毒肽。
CGG重复扩增导致FMR1(NM_002024.6:c- 128GGC[200])5’UTR高甲基化和基因沉默。这导致神经元发育所需的FMR1蛋白量不足,从而引起脆性X综合征。GIPC1 5’UTR中的CGG重复扩增与眼咽远端型肌病(oculopharyngodistal myopathy,OPDM)有关。对照组中CGG重复的数量<30,而在受影响的个体中>60。然而,基因对重复次数的敏感性可以更高;在PTCH1 5’UTR(NM_000264.5:c.- 4dup)中插入多余的第8个CGG三联体,与野生型7次重复序列相比,会极大地抑制蛋白质翻译。据报道,这种非编码变异会增加基底细胞癌的易感性。
在3’UTRs中存在多个致病性重复扩展实例。例如,DMPK的3’UTR中存在一个大的CTG重复(NM_004409.5:c.*224CTG[330]),通过毒性的功能获得机制导致强直性肌营养不良1型(DM1)。有假设认为,突变DMPK转录本形成异常结构并异常地与RBPs结合。同样,脊髓小脑性共济失调8型(SCA8)可由ATXN8OS的3’UTR中的CTG扩张(NR_002717.2:h.1103CTG[107_127])引起。在此,有关于毒性RNA和毒性蛋白质效应的证据,因为RAN翻译可以从反义链生成聚谷氨酸蛋白。
伯科设计合成的人全外显子Panel对多个基因的UTR区域进行了加强覆盖,搭配非编码区致病位点补充Panel,可以对UTR区域进行更全面的检测,保证非翻译区变异的深入研究。
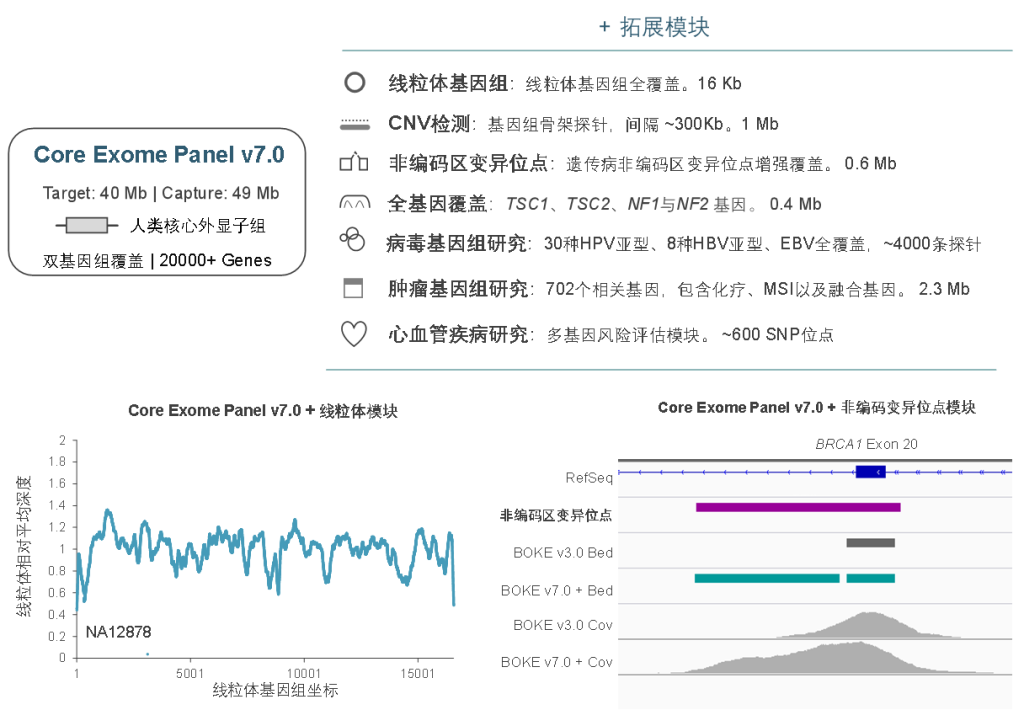
伯科全外芯片 – Core Exome Panel v7.0
Core Exome Panel v7.0(下文简称BOKE v7.0),该WES Panel增强了基因组hg19传统研究区域的覆盖,兼顾 hg19 & hg38 双版本基因组,可以更好的保证临床科研与转化的延续性。目标区域和捕获区域大小分别为40Mb和49Mb,对 hg19 传统研究区域覆盖提升至99.7%(友商I-v1),hg38传统研究区域覆盖相近(友商A-v8)。同时,新添加数百个具有一定功能与表型的基因,总基因数量达到20000+。
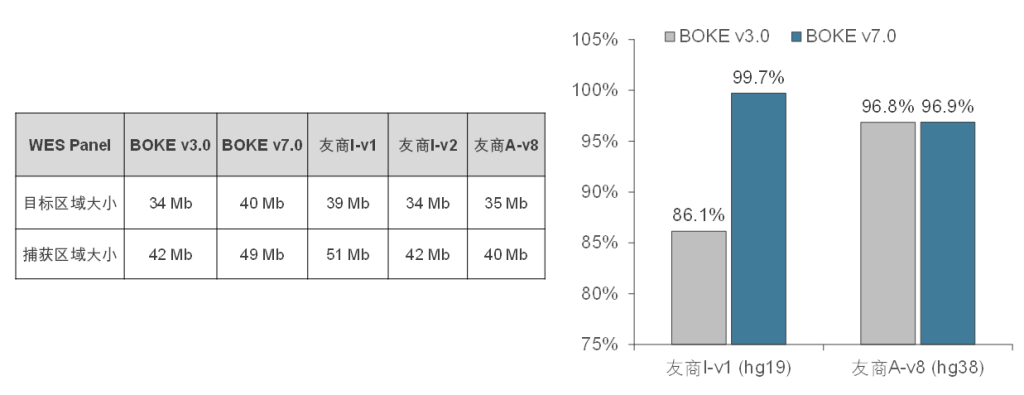
BOKE Core Exome Panel v7.0目标区域大小以及对不同友商产品目标区域的覆盖情况
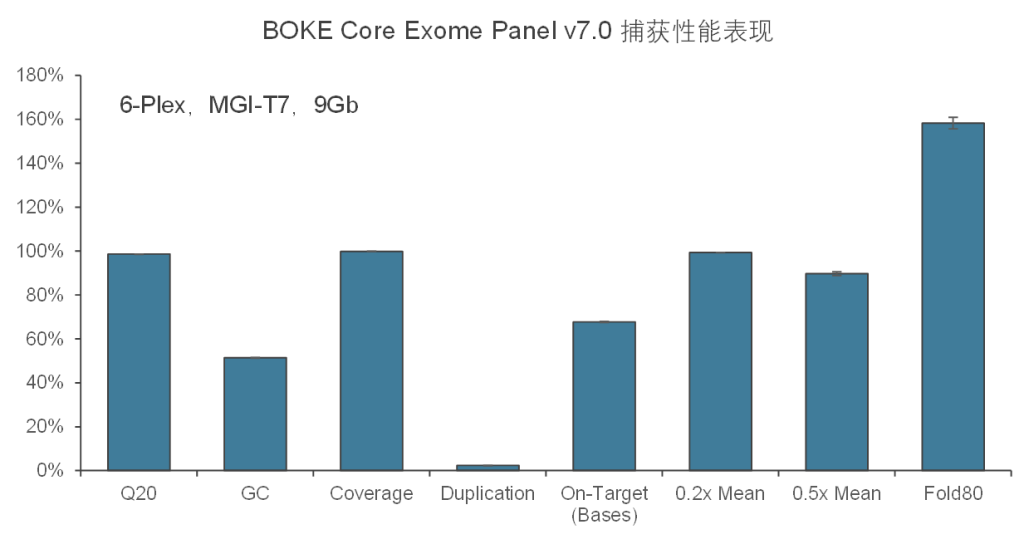
在捕获性能方面,BOKE Core Exome Panel v7.0依然表现优异,与BOKE Core Exome Panel v3.0表现相近。在测序9Gb条件下,平均深度达到110x左右,20x和30x以上区域占比分别为99.5%和98.5%,Fold 80为1.5-1.6之间,与国际领先产品数据表现相当。
同时,BOKE Core Exome Panel v7.0也可灵活的与拓展模块组合使用,满足不同场景的临床研究的需求及转化应用,包括线粒体、遗传病非编码区变异位点、单基因全覆盖、病毒基因组、肿瘤全景变异检测、重大疾病多基因风险评估模块等。此外,伯科公司自研自造的寡核苷酸合成平台可以快速响应个性化定制的需求,为人类基因组分子遗传学的研究与转化,提供更加全面高效的解决方案。
参考资料
The role of untranslated region variants inMendelian disease: a review. European Journal of Human Genetics. 2025