


线粒体疾病:从分子机制到治疗进展
- boke
- 2025-10-31
- 5:01 下午
线粒体疾病是一类常见的遗传性疾病,具有显著的表型和遗传异质性,临床症状可累及全身多个系统和器官,严重程度与表现形式各不相同。线粒体遗传性疾病可由线粒体 DNA(mtDNA)或核 DNA(nDNA)的多种突变引起,这些 DNA 编码线粒体蛋白或其他线粒体组分。
由于线粒体与线粒体疾病之间的关系复杂,人们对这类疾病的基因型 – 表型关联认识不足,历史上其诊断和治疗难度较大,临床预后往往不理想。然而,近年来的研究与技术进展显著提升了我们对这类疾病的认知和管理水平,极大的推动了线粒体相关疗法的临床转化。
线粒体疾病的本质
由线粒体 DNA(mtDNA)或核 DNA(nDNA)突变引起,突变导致线粒体功能 / 代谢紊乱,进而引发能量供应不足及下游病理反应。
线粒体疾病的特征
遗传与表型异质性极强,可累及全身(尤其脑、神经、眼、心脏、骨骼肌等高耗能组织),临床症状多样(发育迟缓、癫痫、肌病、视力障碍等),诊断与治疗难度大。
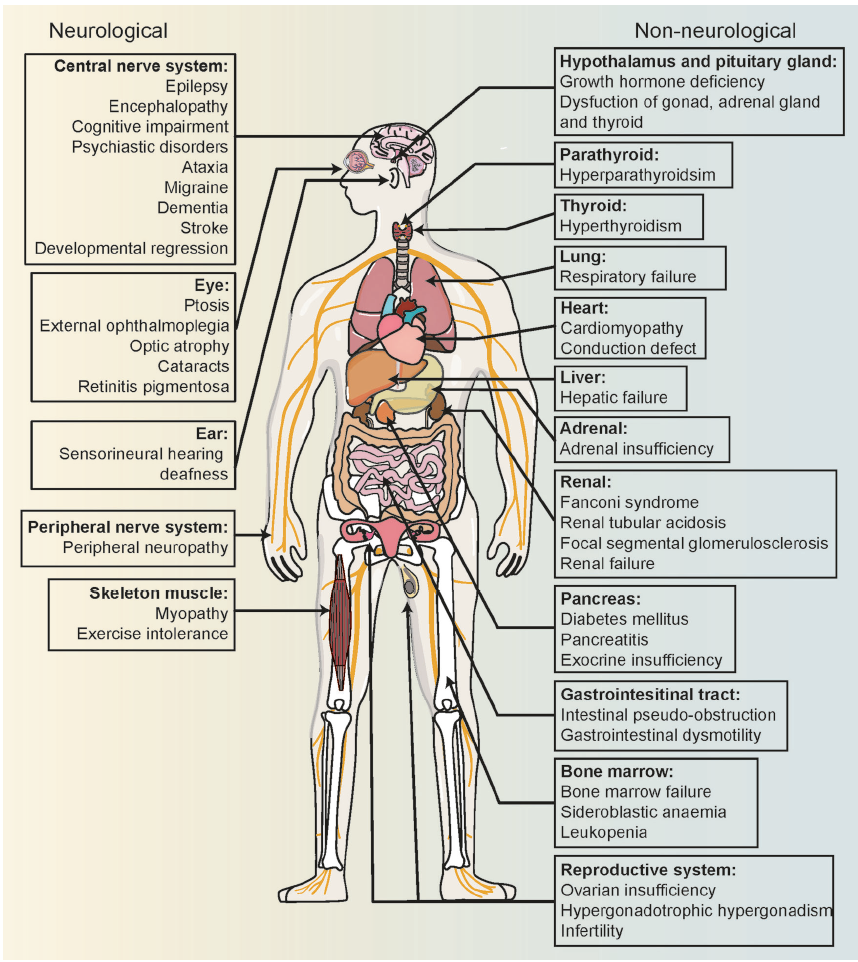
Multisystem Clinical Presentation of Mitochondrial Diseases.
线粒体疾病研究里程碑
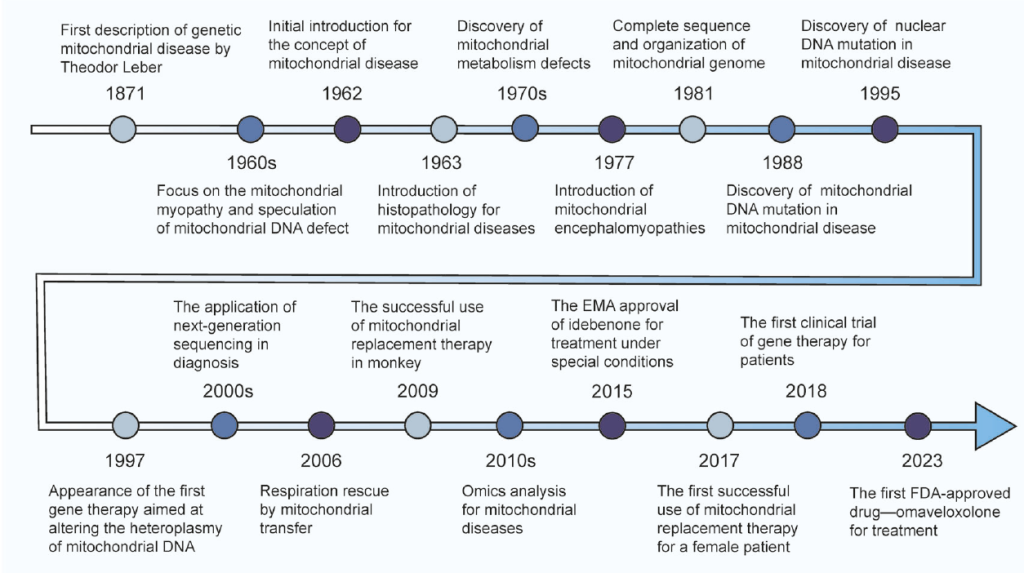
Timeline of Major Historical Events in the Study of Mitochondrial Diseases.
随着对线粒体认知的深入,人们对线粒体遗传性疾病的突变基因和发病机制的了解也不断加深。除了原发性或继发性氧化磷酸化缺陷,影响线粒体结构和功能的基因突变也成为研究重点。
线粒体疾病的分子机制
线粒体疾病的特征是线粒体功能或结构的原发性或继发性缺陷,由 nDNA 或 mtDNA 突变引起 。具体而言,36 个致病基因(11%)由 mtDNA 编码,302 个(89%)由 nDNA 编码。大多数健康人存在 mtDNA 异质性,但不会引发线粒体疾病;只有当突变型 mtDNA 比例超过特定阈值时,才会出现症状。由于显著的遗传和临床异质性,线粒体疾病可影响单个或多个器官系统,临床表现广泛。
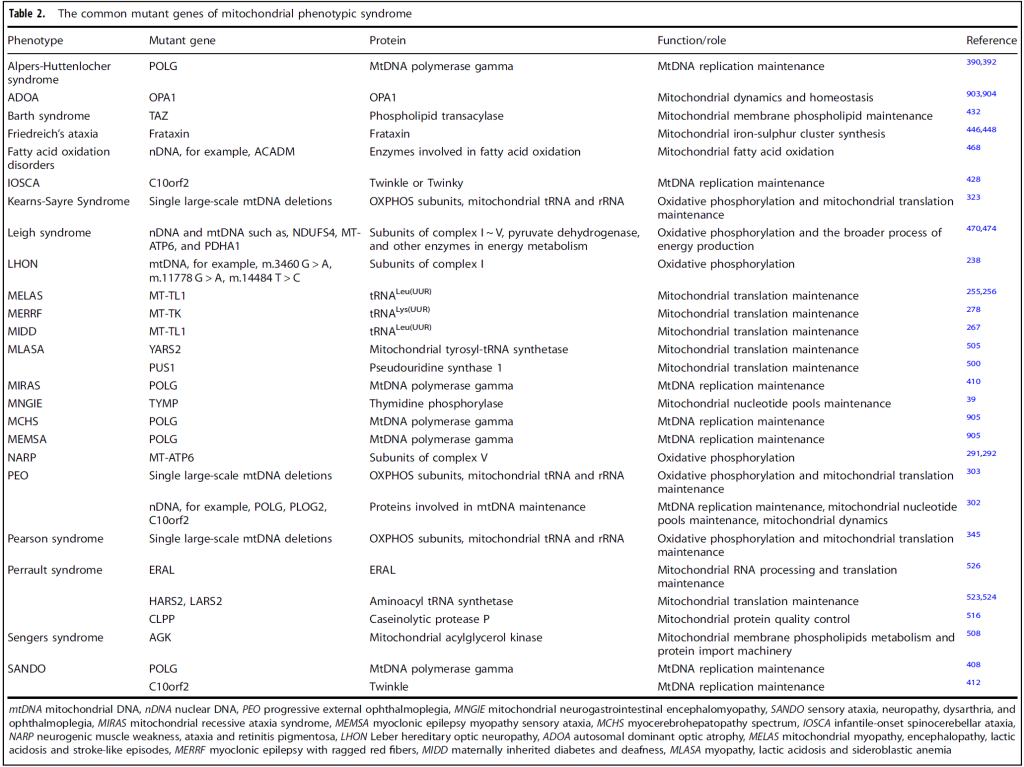
一、mtDNA 突变导致的原发性线粒体疾病
mtDNA 突变导致的线粒体疾病,其典型特征是氧化磷酸化的原发性紊乱,通常引发能量不足、氧化应激增加和 Δ ψ m 崩溃。
1.Leber 遗传性视神经病变(LHON)
主要由 mtDNA 中 MT-ND1(m.3460 G>A)、MT-ND4(m.11778 G>A)、MT-ND6(m.14484 T>C)这三种点突变引起(占比超 95%),呈母系遗传,男性发病率显著更高。
核心表现为视网膜神经节细胞变性,导致双侧严重中心视力丧失;关键机制是复合体 I 功能缺陷引发线粒体活性氧(mtROS)过量,进而诱导视网膜神经节细胞凋亡,且无髓鞘轴突对能量不足尤为敏感。
2.线粒体肌病、脑病、乳酸酸中毒和卒中样发作(MELAS)
多由 mtDNA 中 MT-TL1 基因的点突变导致,其中 m.3243 A>G 突变占比超 80%,m.3271 T>C 突变占 7%-15%,呈母系遗传。
典型症状包括乳酸酸中毒、伴恶心呕吐的头痛、癫痫、卒中样发作,还可能合并耳聋、糖尿病、心肌病等。
发病机制是 MT-TL1 突变致线粒体转运 RNA(mt-tRNALeu (UUR))氨酰化缺陷,破坏线粒体翻译过程,进而损害呼吸链复合体 I 和 IV 的合成。
3.母系遗传性糖尿病伴耳聋(MIDD)
约 85% 病例由 mtDNA 中 MT-TL1 基因的 m.3243 A>G 点突变引起,呈母系遗传。
核心症状为糖尿病与双侧感音神经性听力丧失,部分患者还会出现肌病、神经病变、肾病等并发症;发病与 mtDNA 异质性相关(MIDD 患者异质性通常 <45%,而同一突变在异质性> 85% 时多表现为 MELAS)。
机制是胰腺 β 细胞线粒体功能障碍导致 ATP 生成减少、ROS 增加,最终使胰岛素分泌逐渐不足。
4.肌阵挛性癫痫伴破碎红纤维(MERRF)
超 80% 病例由 mtDNA 中 MT-TK 基因的 m.8344 A>G 点突变引起,少数与 MT-RNR1、MT-RNR2 基因突变相关,呈母系遗传。
主要表现为进行性肌阵挛性癫痫,肌肉活检可见 “破碎红纤维”,还可能合并共济失调、心肌病、痴呆等。
关键机制是 MT-TK 突变阻碍 mt-tRNALys (UUR) 第 58 位的 N¹- 甲基腺苷修饰,导致线粒体翻译异常,破坏呼吸链复合体 I 和 IV 的功能。
5.神经源性肌萎缩、共济失调和视网膜色素变性综合征(NARP)
由 mtDNA 中 MT-ATP6 基因的 m.8993 T>G 或 m.8993 T>C 点突变引起,呈母系遗传。
症状包括肌萎缩、感觉神经病变、共济失调、视网膜色素变性、癫痫等,表型受突变异质性影响 —— 异质性超 85% 多表现为儿童期发病的 Leigh 综合征,60%-70% 时多为成人期发病的 NARP,70%-85% 时两种疾病均可能发生。
机制是突变损害 ATP 合酶(复合体 V)的质子转运功能,导致氧化磷酸化紊乱。
6.进行性眼外肌麻痹(PEO/CPEO)
mtDNA 相关类型多为散发性单一大片段缺失(非母系遗传),由早期胚胎发育过程中 mtDNA 自发突变导致。
核心症状为进行性双侧上睑下垂、弥漫性对称性眼外肌麻痹,可分为单纯型(孤立发病)、Kearns-Sayre 综合征(KSS)、PEO-plus 综合征(伴肌病或其他眼外症状)三类。
机制是 mtDNA 缺失损害氧化磷酸化相关蛋白功能,导致能量生成不足。
7.Kearns-Sayre 综合征(KSS)
由 mtDNA 单一大片段缺失引起,以 4977bp “常见缺失” 最典型,呈散发性(源于早期胚胎 mtDNA 自发突变)。
20 岁前发病,核心为 “视网膜色素变性 + PEO + 完全性心脏传导阻滞” 三联征,还可能合并小脑共济失调、脑脊液蛋白升高、耳聋、糖尿病等。
机制是 mtDNA 缺失导致呼吸链蛋白及翻译必需 tRNA 丢失,引发 ATP 生成不足与氧化应激增加。
8.Pearson 综合征
由 mtDNA 单一大片段缺失引起,4977bp 缺失占 40%-50% 病例,呈散发性。属致命性多系统疾病,首发症状为铁粒幼细胞性贫血,还可能合并胰腺外分泌功能不全、乳酸酸中毒、颅内出血、先天性畸形,预后极差(0-15 岁患者平均死亡年龄 5.44 岁)。
机制是 mtDNA 缺失致氧化磷酸化缺陷,且可能随年龄进展为 KSS(因组织特异性 mtDNA 缺失负荷变化)。
二、核 DNA(nDNA)突变导致的原发性线粒体疾病简述
由于绝大多数线粒体蛋白由 nDNA 编码,nDNA 突变引发的线粒体疾病不仅包括氧化磷酸化紊乱,还涉及线粒体 DNA(mtDNA)维持、线粒体功能与结构及线粒体 – 核通讯所必需的各类结构或功能蛋白缺陷。因此,nDNA 突变相关线粒体疾病的发病机制比 mtDNA 突变相关疾病更为复杂。
1.常染色体显性视神经萎缩(ADOA)
通常 20 岁前发病,核心表现为进行性双侧视力丧失和色觉障碍,主要病理是视网膜神经节细胞(RGCs)变性脱髓鞘;ADOA-plus 综合征还伴多系统症状。
最常见致病基因为 OPA1(编码线粒体内膜融合相关 GTP 酶),OPA3、Drp1、Mfn2 等基因也可能致病。
发病机制主要是 OPA1 突变致线粒体融合障碍,引发线粒体碎片化、轴突转运 “拥堵”,且过度自噬与氧化应激加剧 RGCs 损伤,还可能影响线粒体 – 核通讯及细胞间线粒体转移。
2.Alpers-Huttenlocher 综合征
常染色体隐性遗传的早发肝脑综合征,典型表现为 “顽固性癫痫 + 发育倒退 + 肝功能障碍” 三联征,丙戊酸盐会加重肝损伤,属禁忌。
主要由 POLG 基因突变(如 p.A467T)导致 mtDNA 耗竭,C10orf2 复合杂合突变也可能致病;肌脑肝病变谱综合征(MCHS)是其早期或较轻表型,可能进展为此病。
机制是 POLG 突变破坏 mtDNA 维持,致神经元与星形胶质细胞线粒体功能崩溃,星形胶质细胞神经毒性增强,目前无有效治疗,仅能对症支持。
3.共济失调 – 神经病变谱综合征
涵盖多种疾病,包括感觉性共济失调、神经病变、构音障碍和眼肌麻痹(SANDO)、线粒体隐性共济失调综合征(MIRAS)、婴儿期发病的脊髓小脑共济失调(IOSCA)等。
SANDO 主要由 POLG 隐性突变致肌肉 / 神经组织 mtDNA 多重缺失,C10orf2、RNASEH1 突变也可能致病;MIRAS 为成人发病,伴枕叶癫痫,与 POLG 突变相关;IOSCA 由 C10orf2 突变致 mtDNA 耗竭。
核心机制是相关基因缺陷破坏 mtDNA 复制 / 修复,引发线粒体功能障碍,小脑组织自噬受损是共济失调关键,目前仅能支持对症治疗。
4.Barth 综合征
X 连锁隐性线粒体疾病,男性发病,核心表现为 “肥厚型心肌病 + 骨骼肌病 + 中性粒细胞减少” 三联征,伴线粒体嵴与呼吸链异常。
致病基因为 TAZ(编码酰基特异性磷脂转移酶),突变导致线粒体内膜四亚油酰心磷脂缺乏、单溶血心磷脂积累,破坏呼吸链超复合体稳定性,还阻碍自噬启动。
治疗重点是管理心力衰竭、心律失常与中性粒细胞减少,物理治疗和营养支持辅助,以恢复心磷脂为目标的脂质替代疗法具有前景。
5.弗里德赖希共济失调(Friedreich’s ataxia)
最常见的常染色体隐性脊髓小脑共济失调,25 岁前发病,表现为进行性肢体 / 躯干共济失调,伴构音障碍、深感觉丧失、脊柱侧弯和心肌病。
致病原因是 FXN 基因(X25)第一内含子 GAA 三核苷酸重复不稳定扩增,少数为复合杂合突变,导致 Frataxin 蛋白减少。
机制是 Frataxin 缺乏致铁硫(Fe-S)簇组装障碍,呼吸链复合体 I/II/III 功能缺陷,线粒体铁积累引发严重氧化应激,可能涉及铁死亡,果蝇模型显示线粒体转运受损也参与发病。
6.脂肪酸氧化障碍
常染色体隐性遗传综合征,最常见类型为 ACADM 基因突变导致的中链酰基辅酶 A 脱氢酶缺乏。临床以 “代谢危机”(非酮症性低血糖、呕吐、脑病、酸中毒)和 “肌肉症状”(横纹肌溶解症、运动不耐受)为核心。
机制是相关基因突变致脂肪酸 β- 氧化缺陷,ATP 生成不足,且积累的脂肪酸与肉碱衍生物具有脂毒性,抑制呼吸链、解偶联氧化磷酸化、引发 mPTP 持续开放,最终导致细胞死亡。
7.Leigh 综合征(亚急性坏死性脑病)
儿童最常见线粒体疾病,2 岁前发病,表现为肌张力低下、癫痫、呼吸窘迫、神经发育迟缓、共济失调、乳酸酸中毒。
致病基因超 75 个,nDNA 基因如 NDUFS4(复合体 I 缺陷主因)、PDHA1(丙酮酸脱氢酶缺陷)、SUCLA2/SUCLG1(mtDNA 耗竭相关),mtDNA 突变也可能致病。
机制是氧化磷酸化核心组件缺陷,能量供应崩溃,线粒体质量控制机制异常参与发病;有趣的是,疾病模型中缺氧可通过减少 mtROS、激活缺氧诱导因子缓解症状。
8.线粒体神经胃肠脑肌病(MNGIE)
常染色体隐性疾病,表现为恶病质、胃肠道动力障碍(恶心、呕吐、排便异常)、周围神经病、眼肌麻痹和脑白质病。
主要致病基因为 TYMP,突变导致胸苷磷酸化酶活性丧失,RRM2B、LIG3 突变也可能引发类似表型。
机制是 TYMP 突变致脱氧胸苷 / 脱氧尿苷积累,破坏线粒体核苷酸库平衡,mtDNA 复制异常(点突变、多重缺失、耗竭),且核苷积累改变溶酶体酸性环境,阻碍缺陷线粒体降解。
治疗包括血液透析、造血干细胞移植(HSCT)等,基因治疗在小鼠模型中已显效。
9.肌病、乳酸酸中毒和铁粒幼细胞性贫血(MLASA)
常染色体隐性疾病,影响骨骼肌与骨髓,核心表现为线粒体肌病、乳酸酸中毒和铁粒幼细胞性贫血。
主要致病基因为 PUS1(编码假尿苷合酶 1),YARS2 基因突变也可致病。机制是 PUS1 突变致 mt-tRNA 假尿苷化修饰缺陷,YARS2 突变降低线粒体酪氨酰 – tRNA 合成酶氨酰化活性,二者均引发线粒体翻译异常,最终导致氧化磷酸化崩溃,红细胞生成受破坏。
10.Sengers 综合征
常染色体隐性线粒体疾病,表现为肥厚型心肌病、骨骼肌病、先天性白内障和乳酸酸中毒。
致病基因为 AGK(编码线粒体酰基甘油激酶)。
机制是 AGK 突变不仅影响线粒体内膜心磷脂合成,还破坏其作为 TIM22 复合体亚基的功能(阻碍线粒体载体蛋白导入),可能进一步干扰呼吸链复合体 I 功能,具体分子机制仍需深入研究。
11.Perrault 综合征
常染色体隐性疾病,女性核心表现为 “感觉神经性听力丧失 + 卵巢发育不全”,男性多仅听力丧失,部分伴神经系统症状。
致病基因包括 CLPP、ERAL1、HARS2、LARS2、C10orf2 等。机制是 CLPP 突变致线粒体蛋白酶解活性下降,HARS2/LARS2 突变影响 mt-tRNA 氨酰化,ERAL1 突变阻碍线粒体核糖体组装,最终均导致线粒体蛋白稳态失衡。
治疗以对症为主,如人工耳蜗植入(改善听力)、雌激素替代疗法(改善卵巢发育),有生育需求者可考虑体外受精。
年龄相关疾病与线粒体的关联
1.核心背景与线粒体功能变化
衰老会导致线粒体功能渐进性下降,具体表现为氧化磷酸化(OXPHOS)效率降低、ATP 生成减少、活性氧(ROS)产生增加,同时伴随 mtDNA 突变(如点突变、多重缺失)与拷贝数异常(AD 中 mtDNA 突变和拷贝数减少,PD 中 mtDNA 缺失显著)、线粒体质量控制(MQC)网络紊乱。这些变化会加速氧化产物积累,且死亡神经元释放的 mtDNA、碎片化线粒体等物质还会启动炎症反应,共同推动阿尔茨海默病(AD)、帕金森病(PD)等神经退行性疾病的发生发展。
2.与阿尔茨海默病(AD)的关联
AD 中神经元损伤的核心机制是 “病理性蛋白聚集→氧化应激→线粒体功能障碍→神经炎症” 的恶性循环。β- 淀粉样蛋白积累与 tau 蛋白聚集会直接加剧氧化应激,进而破坏线粒体生物能量学(如呼吸链功能)和 MQC 系统;同时,线粒体与自噬功能障碍会激活小胶质细胞,引发神经炎症,进一步加重神经元损伤,形成病理进展闭环。
3.与帕金森病(PD)的关联
PD 的线粒体相关病理主要涉及两方面:一是 α- 突触核蛋白在线粒体膜形成寡聚体,抑制呼吸链复合体 I 活性,导致 mtROS 过量产生,最终诱导多巴胺能神经元凋亡;二是 PINK1 或 Parkin 基因突变会破坏不依赖 PINK1-Parkin 通路的自噬功能,导致缺陷线粒体无法有效清除而大量积累。此外,PD 相关神经毒素和基因突变还会诱导线粒体异常分裂,进一步加剧神经炎症,推动疾病进展。
4.其他影响因素与潜在靶点
除衰老和线粒体功能障碍外,生活方式(如饮食、运动)、环境暴露(如神经毒素),以及基因组不稳定性、端粒磨损、表观遗传修饰、营养感知失调等,均会增加神经退行性疾病易感性。
5.与遗传性线粒体疾病的差异
尽管两类疾病均以线粒体功能障碍为核心特征,但存在显著区别:年龄相关疾病是病理性蛋白聚集、MQC 紊乱等因素长期积累的结果,发病年龄较晚(多为中老年);遗传性线粒体疾病由 mtDNA 或 nDNA 特定突变直接损害线粒体功能导致,发病早(多为婴儿期或青少年期),临床表型更集中于多系统损伤(如肌病、脑病)。
线粒体疾病的诊断方法
由于线粒体遗传疾病存在遗传和表型异质性,准确诊断面临巨大挑战。除细致的临床观察外,还需进行全面检查,包括体液生化分析、神经影像学检查、DNA 和 RNA 测序,以及组织生化或病理学检查。
怀疑线粒体疾病时,应首先对血液、尿液和脑脊液进行生化检测;然而,尚无完美的诊断方法;诊断非典型和新型线粒体遗传疾病,需审慎结合多种诊断手段。此外,必须强调临床观察与实验室检查结合的重要性 —— 这种关联是准确诊断的关键。
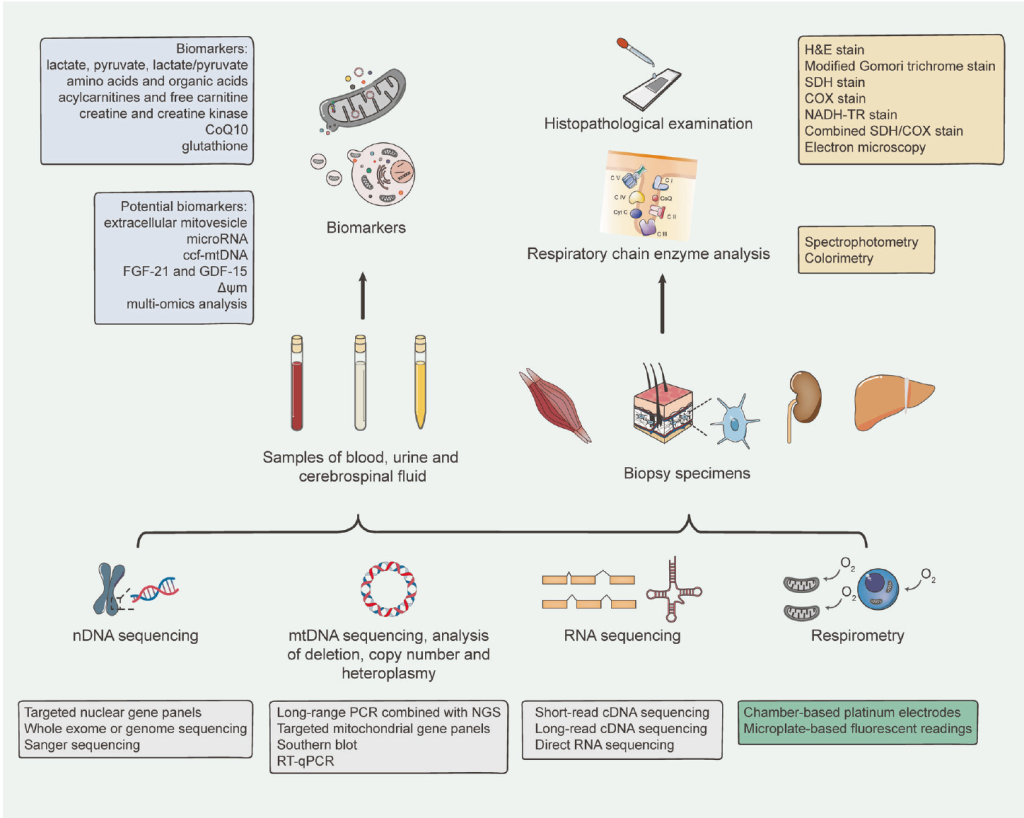
Diagnostic Methodology for Mitochondrial Diseases.
(一)生物标志物
代谢标志物:乳酸 / 丙酮酸(特异性 83%-100%,乳酸 / 丙酮酸比值区分 PDHC 缺陷)、FGF-21/GDF-15(线粒体肌病诊断敏感性高);
分子标志物:循环无细胞 mtDNA(ccf-mtDNA,MELAS 特异性 94%)、mitoEVs(脑病指标)、miRNA(miRNA-27b-3p 用于 MELAS)。
(二)基因与转录组测序
1.mtDNA 检测
由于 mtDNA 突变是线粒体遗传疾病的主要病因,诊断过程中应优先进行 mtDNA 检测。下一代测序(NGS)技术的出现,使诊断策略从 “活检优先” 转向 “遗传优先”(一线方法);随着 NGS 的发展和广泛应用,这类疾病的遗传诊断率已从 NGS 前时代的 10%-20% 提升至如今的 40%-60%。
近期,英国发布了新的线粒体疾病遗传诊断实践指南,旨在规范和指导 DNA 测序的应用。
NGS需分析全基因组、缺失、拷贝数、异质性。
血液 / 尿液 / 受累组织联合检测(避免组织特异性突变遗漏)。
2.nDNA 检测
采用靶向基因 panel 和全外显子测序对参与 mtDNA 维持、线粒体功能和代谢的核基因进行 NGS 检测,是首选方法;儿童患者优先。
3.RNA 测序
补充基因组测序,可帮助诊断基因组测序后仍未明确遗传病因的疑似线粒体疾病:检测异常剪接(如 CLPP 突变与 Perrault 综合征)、tRNA 缺陷。
(三)活检与功能评估
组织活检:肌肉活检(金标准)检测破碎红纤维、COX/SDH 染色;皮肤活检分析成纤维细胞;
生化分析:测定呼吸链酶活性(分光光度法)、CoQ10 水平;
呼吸测定法:高分辨率 respirometry 评估 OXPHOS 功能,底物 – 解偶联剂 – 抑制剂滴定(SUIT)定量复合体活性。
线粒体疾病的治疗策略
大多数线粒体疾病的有效治疗方法仍有待探索,但线粒体置换疗法(MRT) 和基因治疗是治疗线粒体遗传疾病及预防其向后代传递的有前景基础方法;此外,恢复线粒体 tRNA 转录后修饰、开发特定疾病靶向疗法的研究也在不断推进。
线粒体遗传疾病的临床试验中,MRT、基因治疗、药物治疗、细胞治疗等已显治疗潜力,多通过替换缺陷核酸、抗氧化、调控代谢等机制起效;虽广泛应用存挑战,但可借基因编辑、个性化方案、高效递送系统等克服。此外,间充质干细胞线粒体转移、缺氧干预等潜在疗法待深入研究以推进临床,诱导多能干细胞与类器官技术也为探究发病机制和治疗策略提供有力支撑。
(一)线粒体置换疗法(MRT,预防遗传传递)
原核移植(PNT):转移受精卵原核至健康去核卵母细胞,英国获批;
纺锤体 – 染色体复合体转移(ST):转移 MII 卵母细胞纺锤体,已诞健康婴儿;
极体移植(PBT):利用极体低线粒体特性,降低供体 mtDNA 残留;
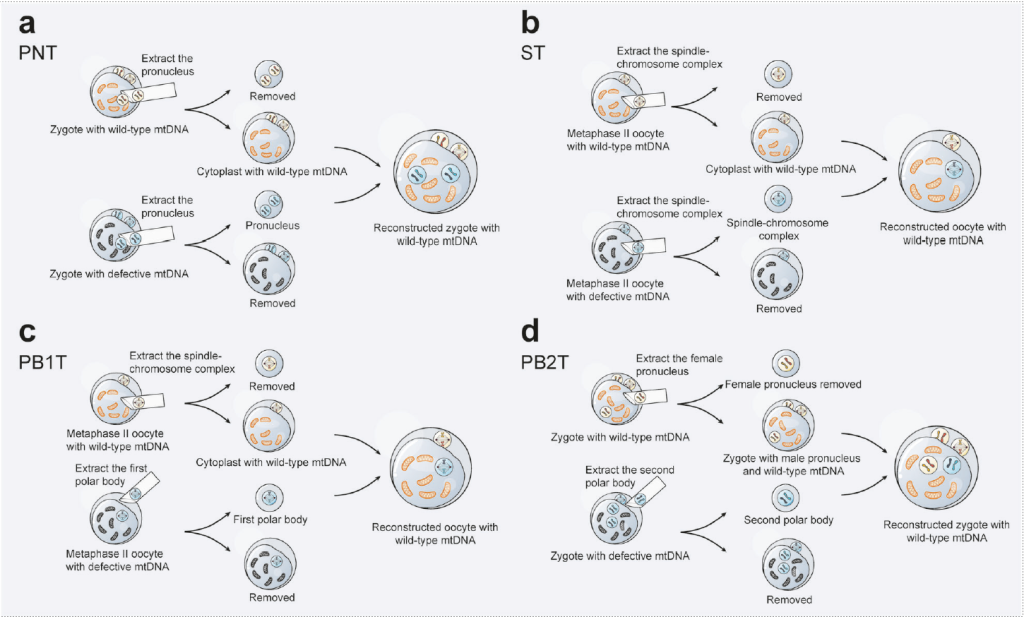
Procedure for Mitochondrial Replacement Therapy.
(二)基因治疗(纠正突变)
1.核基因治疗:
异源表达:将 mtDNA 基因(如 MT-ND4)重新编码后导入细胞核,AAV 介导递送至 RGCs(LHON 临床试验);
基因替代:AAV 递送正常 nDNA(如 Frataxin、TAZ),改善弗里德赖希共济失调、Barth 综合征;
2.mtDNA 编辑:
核酸酶:mtZFNs、mitoTALENs、mitoARCUS 清除突变 mtDNA;
碱基编辑:DdCBEs 实现 C-to-T/A-to-G 编辑,纠正点突变(如 m.3243 A>G);
挑战:CRISPR/Cas9 因线粒体 RNA 转运障碍暂难应用。
(三)药物治疗(对症 / 改善功能)
抗氧化剂:CoQ10(增强电子传递)、艾地苯醌(LHON 治疗)、EPI-743(改善谷胱甘肽);
代谢调节剂:奥马韦洛酮(激活 Nrf2,弗里德赖希共济失调)、苯扎贝特(激活 PPAR,诱导线粒体生物发生);
螯合剂:去铁酮(清除线粒体铁,弗里德赖希共济失调);
其他:L – 精氨酸(MELAS 预防卒中样发作)、脱氧核苷(TK2 缺陷)。
(四)其他疗法
细胞治疗:中胚层血管母细胞移植(补充野生型 mtDNA)、造血干细胞移植(MNGIE 恢复 TP 活性);
酶替代治疗:红细胞包裹 TP(EE-TP,MNGIE);
器官移植:肝移植(MNGIE、乙基丙二酸脑病)、心脏移植(KSS);
运动 / 康复:中等强度有氧运动促进线粒体增殖,低强度运动增强基因治疗递送;
iPSCs 与类器官:建模疾病(如 MELAS、Leigh 综合征)、筛选药物、探索细胞治疗。
结论与未来展望
(一)现有进展
机制认知:从 “能量缺陷” 拓展至 “通路性疾病”,MQC、线粒体 – 核通讯、炎症的作用获重视;
诊断突破:NGS 使遗传诊断率从 10%-20% 提升至 40%-60%,多组学(蛋白组 / 脂质组)助力生物标志物发现;
治疗潜力:MRT、AAV 基因治疗、碱基编辑进入临床,药物从对症向病因治疗迈进。
(二)核心挑战
基因型 – 表型关联不明,疾病模型(尤其组织特异性)不足;
mtDNA 编辑效率与脱靶风险、MRT 核 – 线粒体不相容待解决;
多数疾病仍无根治手段,治疗窗口窄、个体差异大。
(三)未来方向
深入线粒体 – 核通讯(如乳酸化表观调控)、组织特异性 MQC 机制;
开发更精准的基因编辑工具(低脱靶、易递送)、多疗法联合(如基因治疗 + 药物);
利用 iPSCs / 类器官实现个性化治疗,探索体外卵母细胞生成降低 MRT 供体需求。
核心观点总结
线粒体疾病是一类由 mtDNA/nDNA 突变引发的 “通路性疾病”,其复杂性源于线粒体功能的多效性与异质性;诊断上可整合多组学提升准确性,应对测序诊断率不稳与意义未明变异(VUS)挑战;基因治疗需突破递送限制,开发精准、低脱靶的工具,也可结合 MRT 增强疗效,药物与运动疗法需依临床证据,联合治疗或有协同益处;还应扩大 iPSCs 和类器官应用,用于疾病建模、新疗法开发,探索其分化为卵母细胞以助力 MRT,推动精准诊断与有效治疗的临床转化。
参考文献:
1.Haipeng Wen, et al. Mitochondrial diseases: from molecular mechanisms to therapeutic advances. Signal Transduction and Targeted Therapy (2025) 10:9; https://doi.org/10.1038/s41392-024-02044-3