


消化道肿瘤精准治疗:靶点、机制、药物与展望(下)
- boke
- 2025-11-18
- 5:04 下午
胃癌特异性靶点
一
CLDN18.2(紧密连接蛋白 Claudin 18 亚型)
1. 生物学意义:维持细胞旁屏障功能,高表达时促进肿瘤侵袭,正常胃黏膜特异性表达,肿瘤中异常高表达,是胃癌精准治疗的核心靶点 [12,13]。

2. 异常特征:
表达率:胃癌中高表达率 35%-46.5%,其中弥漫型胃癌 48.3%、肠型胃癌 38.8%[12];
阳性定义更新:≥40% 肿瘤细胞膜染色强度≥2+;靶点依赖性:CLDN18.2 高表达(≥70%+)患者的 mPFS 显著优于中低表达(7.1 个月 vs 4.2 个月,P=0.00257)[12,13];
挑战:潜在脱靶效应、表达异质性 [12,13];
3.分子机制:
通过维持细胞间紧密连接完整性,增强肿瘤细胞侵袭能力,同时调控肿瘤微环境免疫抑制 [12,13]。
4.靶向药物研发及临床数据
佐妥昔单抗(Zolbetuximab)
机制:嵌合 IgG1 单抗,特异性结合 CLDN18.2,通过 ADCC、CDC 效应杀伤肿瘤细胞 [12,13]。
临床试验:Ⅱ 期 FAST 随机试验、Ⅲ 期试验 [12,13]。
临床数据:联合 mFOLFOX6 化疗,CLDN18.2 高表达(IHC 2+/3+≥75%)患者,mPFS 10.6 个月,mOS 18.2 个月,显著优于单纯化疗(PFS 8.6 个月,OS 15.5 个月);主要不良反应为恶心(64%)、呕吐(57%),多为 1-2 级 [12,13];Ⅱ 期 FAST 随机试验显示,佐利妥昔单抗联合化疗相较于单纯化疗,可显著改善 Claudin 18.2 阳性晚期胃癌 / 胃食管交界腺癌患者 OS 和 PFS [12]。
获批情况:2023 年欧盟批准用于 CLDN18.2 高表达晚期胃癌 [12,13]。
CAR-T 细胞治疗(CT041/satri-cel)
临床数据:Ⅰ 期全人群研究显示,6 个月 OS 率 81.2%,ORR 57.1%;首次验证 “一线治疗期间提前单采 CAR-T 细胞” 的可行性 ——15 例提前单采患者的 mPFS(7.1 个月)、mOS(10.2 个月)数值优于常规单采人群 [12];
获批情况:2020 年 FDA 批准用于胃癌 / GEJA 治疗 [12];
ADC 药物(Zolbetuximab-deruxtecan):优化 linker 与细胞毒性药物,临床前研发中 [12,13];
其他单抗(GC1118):针对 CLDN18.2 的新型单抗,Ⅰ 期试验显示良好耐受性 [12]。
二
EBV 阳性( Epstein-Barr 病毒阳性)
1. 生物学意义:EBV 感染驱动胃癌发生,同时上调 PD-L1/PD-L2 表达,增强免疫原性 [12,13];
2. 异常特征:占胃癌的 9%,特征为 JAK2、ERBB2 扩增及 PD-L1/PD-L2 超表达 [12,13];
3. 分子机制:EBV 基因组整合入宿主细胞,导致原癌基因扩增及免疫检查点分子高表达,形成免疫抑制微环境 [12,13];
4. 靶向药物研发及临床数据:
PD-1/PD-L1 抑制剂:帕博利珠单抗、纳武利尤单抗单药治疗 EBV 阳性胃癌,ORR 约 40%-50%,显著高于普通亚型 [12,13];
联合方案:PD-1 抑制剂联合化疗(如 XELOX),mOS 约 14 个月,优于单纯化疗 [12,13]。
三
FGFR2 扩增 / 融合
1. 生物学意义:FGFR2 是受体酪氨酸激酶,扩增 / 融合后持续激活下游信号,促进肿瘤增殖与血管生成 [12,13];
2. 异常特征:胃癌中发生率约 5%-7%,多见于 Lauren 肠型 [12,13];
3. 分子机制:FGFR2 扩增 / 融合后,无需配体即可激活 PI3K/AKT、MAPK 通路,促进细胞增殖 [12,13];
4. 靶向药物研发及临床数据
Bemarituzumab(FGFR2b 抑制剂)
机制:特异性结合 FGFR2b,阻断信号激活 [12,13];
临床试验:Ⅰ/Ⅱ 期试验 [12,13];
临床数据:联合化疗治疗 FGFR2 扩增胃癌,ORR 约 35%,mPFS 7.4 个月 [12,13];
FGFR-TKI(如 erdafitinib)
在 FGFR2 融合胃癌中显示部分活性,进入 Ⅱ 期试验 [12,13]。
四
tRF-Val(3’tRNA 衍生片段)
1. 生物学意义:tRF-Val 是转运 RNA(tRNA)衍生的小非编码 RNA(sncRNA),调控肿瘤细胞增殖相关基因表达,促进胃癌生长 [12];
2. 异常特征:在胃癌组织中高频高表达,尤其适用于 p53 通路异常的胃癌亚型 [12];
3. 分子机制:tRF-Val 通过结合靶 mRNA,调控细胞周期相关基因(如 Cyclin D1)表达,促进肿瘤细胞增殖 [12];
4. 靶向药物研发及临床数据:临床前数据:体内实验证实,沉默 tRF-Val 可显著抑制裸鼠胃癌移植瘤生长 [12];研发阶段:针对 tRF-Val 的反义寡核苷酸(ASO)处于临床前研发 [12]。
五
临床前靶点[黏蛋白 MUC1/6/17、HGFR(c-Met)]
黏蛋白(MUC1、MUC6、MUC17)
1.生物学意义:黏蛋白是上皮细胞分泌的糖蛋白,异常糖基化和过表达促进肿瘤侵袭与免疫逃逸 [12];
2.异常特征:MUC1 在胃癌中异常糖基化和特异性过表达率约 40%[12];
3.靶向药物研发:抗 MUC1 单抗(如 clivatuzumab tetraxetan)在临床前模型中显示抗肿瘤活性,进入 Ⅰ 期试验 [12]。
HGFR(c-Met)
1.生物学意义:受体酪氨酸激酶,激活后促进肿瘤侵袭性及不良预后 [12];
2.异常特征:胃癌中 HGFR 扩增率约 5%-10%[12];
3.分子机制:HGFR 与 HGF 结合后激活 PI3K/AKT、MAPK 通路,促进肿瘤增殖与转移 [12];
4.靶向药物研发:c-Met 抑制剂(如 capmatinib)联合 EGFR 单抗在临床前模型中显示协同效应,进入 Ⅰ 期试验 [12]。
六
免疫逃逸相关机制及靶点
1. 肿瘤相关抗原(TAA)缺失或改变:导致免疫系统无法识别肿瘤细胞,是胃癌免疫耐药的原因之一 [12];
2. 抗原呈递机制(APM)损伤:如 HLA 分子表达下调,影响 T 细胞识别,相关修复剂处于临床前研发 [12];
3. 肿瘤微环境(TME)免疫抑制:
分泌免疫抑制因子(如 IL-10、TGF-β);
表达免疫检查点(PD-L1、CTLA-4)[12]。
4. 靶向药物研发及临床数据:
PD-1 抑制剂:
纳武利尤单抗(Nivolumab):Ⅲ 期 CheckMate-649 试验显示,一线联合氟尿嘧啶类 + 铂类化疗,胃癌患者 mOS 显著延长(HR=0.71,P<0.001),2021 年 FDA 批准用于晚期胃癌一线治疗 [12,13];
帕博利珠单抗(Pembrolizumab):Ⅱ 期 KEYNOTE-059 显示三线治疗 PD-L1 阳性(CPS≥1)胃癌 ORR 11.6%;KEYNOTE-811 显示联合曲妥珠单抗 + 化疗可提升 HER2 阳性胃癌 ORR,FDA 已批准该方案 [4,12,13]。
PD-L1 抑制剂
阿替利珠单抗(Atezolizumab):Ⅱ 期 PANDA 试验显示联合 FLOT 化疗治疗可切除胃癌,病理缓解率提高 [12,17,18];
度伐利尤单抗(Durvalumab):联合化疗(如 FOLFIRI)作为胃癌二线 / 三线治疗,ORR 显著提升,安全性可控 [12,13]。
CTLA-4 抑制剂
伊匹木单抗(Ipilimumab):单药治疗胃癌疗效不佳,但联合纳武利尤单抗治疗化疗耐药食管胃腺癌,显示临床意义的抗肿瘤活性 [2,12];
曲美木单抗(Tremelimumab):联合度伐利尤单抗 + 化疗作为胃癌二线 / 三线治疗,可延长患者生存期 [12,13]。
免疫联合策略
HER2 靶向 ADC(如德曲妥珠单抗)联合 PD-1 抑制剂(如帕博利珠单抗):Ib/Ⅱ 期 DESTINY-Gastric03 试验(NCT04379596)显示,该方案可提升 HER2 阳性胃癌患者 ORR 至 60% 以上 [12,13];
雷莫西尤单抗(抗 VEGFR2)联合 PD-1 抑制剂(如纳武利尤单抗):临床试验(如 NCT04704934)正在探索,初步显示协同增效潜力 [12,13];
ICIs 联合肿瘤疫苗:胃癌领域,Claudin 18.2 肽疫苗联合 PD-1 抑制剂的临床试验正在设计,旨在增强 Claudin 18.2 阳性胃癌患者的免疫应答 [12]。
过继性免疫细胞治疗:
——CAR-NK 治疗:Ⅱ 期试验(NCT04847466)评估辐照 PD-L1 CAR-NK + 帕博利珠单抗 + N-803 治疗胃癌,无细胞因子风暴风险,实体瘤穿透性优于 CAR-T [12];
——CIK 治疗:胃癌术后辅助治疗可延长无病生存期,联合 DC 细胞治疗晚期胃癌,mOS 与 PFS 优于单纯化疗;局限为缺乏高级别临床证据支持 [12];
肿瘤疫苗:OTSGC-A24 肽疫苗、MG7-DC 疫苗(临床试验阶段)、mRNA 疫苗(激活肿瘤特异性 T 细胞)[12];
溶瘤病毒:重组人 5 型腺病毒,胃癌肝转移患者两疗程 DCR 74%[12];
非特异性免疫调节剂:IL-2 衍生物联合免疫检查点抑制剂(临床试验阶段)[12]。
胰腺癌特异性靶点
一
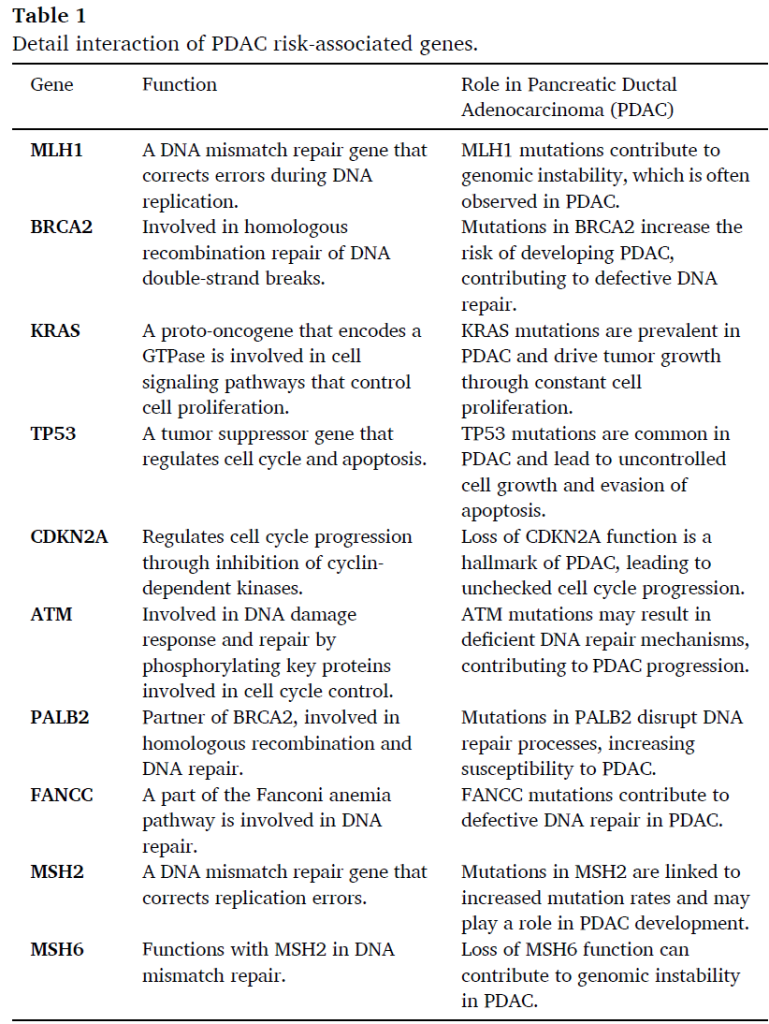
TP53
1. 生物学意义:肿瘤抑制基因,调控细胞周期与凋亡,失活后导致细胞无限增殖 [11];
2. 异常特征:突变率 50%-75%,是胰腺癌第二大高频突变,失活后导致细胞周期失控、DNA 损伤修复缺陷 [11];
3. 分子机制:TP53 正常状态下可识别 DNA 损伤,激活 p21 等细胞周期抑制基因,诱导细胞凋亡;突变后失活,无法发挥抑癌功能,导致细胞恶性增殖 [11];
4. 靶向药物研发:针对 TP53 突变的修复剂(如 PRIMA-1MET)在临床前模型中可恢复突变 TP53 功能,进入 Ⅰ 期试验 [11]。
二
CDKN2A
1. 生物学意义:编码 p16 蛋白,通过抑制 cyclin 依赖性激酶(CDK4/6)调控细胞周期,防止细胞过度增殖 [11];
2. 异常特征:失活率 95%-98%,是胰腺癌的标志性分子异常,失活后导致细胞无限增殖 [11];
3. 分子机制:CDKN2A 失活后,p16 蛋白表达缺失,无法抑制 CDK4/6,导致视网膜母细胞瘤蛋白(Rb)持续磷酸化,细胞周期 G1/S 期检查点失控 [11];
4. 靶向药物研发:
p16 蛋白替代疗法:通过病毒载体递送 p16 基因,临床前模型中可抑制胰腺癌细胞增殖 [11];
CDK4/6 抑制剂:如帕博西尼(Palbociclib),联合化疗在胰腺癌 Ⅱ 期试验中显示部分活性 [11]。
三
SMAD4
1. 生物学意义:TGF-β 通路关键分子,调控细胞分化与凋亡,失活后促进胰腺癌进展 [11];
2. 异常特征:突变率 50%,失活后加速胰腺癌进展 [11];
3. 分子机制:SMAD4 正常状态下可与 SMAD2/3 形成复合物,调控 TGF-β 介导的抑癌信号;突变后失活,TGF-β 信号从抑癌转向促癌,促进 EMT 与转移 [11];
4. 靶向药物研发:TGF-β 受体抑制剂(如 galunisertib)联合化疗在胰腺癌 Ⅱ 期试验中显示协同效应 [11]。
四
BRCA1/2
1. 生物学意义:参与 DNA 同源重组修复,突变后导致 DNA 修复缺陷,对铂类化疗及 PARP 抑制剂敏感 [11];
2. 异常特征:胚系突变率约 5%-7%,体细胞突变率约 10%[11];
3. 分子机制:BRCA1/2 突变后,DNA 双链断裂修复依赖 PARP 介导的单链断裂修复;PARP 抑制剂可阻断该修复途径,导致 “合成致死”[11];
4. 靶向药物研发及临床数据:
PARP 抑制剂(奥拉帕利,Olaparib)
机制:通过抑制 PARP 酶活性,阻断 DNA 单链断裂修复,利用 BRCA1/2 突变细胞的 “合成致死” 效应杀伤肿瘤 [11];
适用瘤种:BRCA1/2 突变、铂类化疗敏感的晚期胰腺癌 [11];
临床试验:POLO(Ⅲ 期)[11];
临床数据:POLO 试验显示,奥拉帕利维持治疗组 mPFS 7.4 个月,显著优于安慰剂组(3.8 个月),OS 有延长趋势但未达统计学差异 [11];
获批情况:2019 年 FDA 批准用于 BRCA1/2 突变、铂类敏感晚期胰腺癌的维持治疗 [11]。
五
NRG1 融合
1. 生物学意义:NRG1(神经调节蛋白 1)与 HER3 形成融合蛋白,持续激活 HER3-PI3K 通路,驱动肿瘤增殖,是 KRAS 野生型胰腺癌的关键驱动靶点 [1,11];
2. 异常特征:发生率约 0.5%-1%,多见于 KRAS 野生型胰腺癌,且常与其他驱动突变互斥 [1,11];
3. 分子机制:NRG1 融合蛋白无需配体即可与 HER3 结合,促进 HER3 二聚化(多与 HER2 形成异二聚体),激活下游 PI3K/AKT/mTOR 通路,促进细胞增殖与存活 [1,11];
4. 靶向药物研发及临床数据:
HER3 抑制剂(Seribantumab)
机制:全人源抗 HER3 单抗,阻断 NRG1-HER3 结合及下游信号 [1,11];
临床试验:Ⅰ/Ⅱ 期试验(NCT02980341)[1,11];
临床数据:在 NRG1 融合胰腺癌患者中,单药 ORR 约 20%,mPFS 5.2 个月,安全性可控(主要不良反应为皮疹、腹泻)[1,11];
双特异性抗体(Zenocutuzumab,MCLA-128)
机制:同时结合 HER2 与 HER3,阻断 NRG1-HER3-HER2 信号轴 [1,11];
临床数据:Ⅰ 期试验中,NRG1 融合胰腺癌患者 ORR 25%,mPFS 6.0 个月,目前进入 Ⅱ 期验证阶段 [1,11]。
六
KRAS 抑制剂(针对胰腺癌 KRAS 突变)
1. 生物学意义:KRAS 是胰腺癌最核心驱动突变(突变率 97.7%),持续激活下游 MAPK/PI3K 通路,是胰腺癌靶向治疗的关键靶点 [11];
2. 异常特征:以 G12D(约 45%)、G12V(约 30%)突变为主,G12C 突变率极低(<1%)[11];
3. 分子机制:同 “泛消化道肿瘤共性靶点 – KRAS 靶点” 的分子机制,突变后持续激活 MAPK、PI3K、Ral-GEF 三条核心通路 [11];
4. 靶向药物研发及临床数据:
KRAS G12C 抑制剂(索托拉西布、阿达格拉西布)
适用人群:仅针对胰腺癌中罕见的 KRAS G12C 突变患者 ;
临床数据:索托拉西布 Ⅰ 期试验中,胰腺癌 KRAS G12C 突变患者 DCR 83%,mPFS 4.0 个月;阿达格拉西布 Ⅰ 期试验 DCR 79%,mPFS 5.6 个月 [11];
KRAS G12D 抑制剂(MRTX-1133、HRS-4642)
机制:非共价结合 KRAS G12D 突变位点,抑制其活性 [11];
临床试验:Ⅰ/Ⅱ 期试验(针对胰腺癌等 G12D 突变实体瘤)[11];
临床前数据:在胰腺癌 PDX 模型中,MRTX-1133 单药肿瘤抑制率超 60%,联合吉西他滨抑制率达 85%[11];
泛 KRAS 抑制剂(BI 1701963)
机制:SOS1 抑制剂,通过抑制 KRAS 上游激活因子,间接抑制所有 KRAS 突变亚型(包括 G12D/V)[11];
临床数据:联合索托拉西布治疗胰腺癌,Ⅰ 期试验显示 DCR 75%,mPFS 5.8 个月,可延缓耐药 [11]。
胃肠道间质瘤(GIST)特异性靶点
一
KIT
1. 生物学意义:KIT 是受体酪氨酸激酶,正常情况下调控造血干细胞、胃肠道 Cajal 间质细胞的增殖与分化;突变后持续激活,是 GIST 最核心的驱动突变 [19];
2. 异常特征:
突变率:70%,是 GIST 第一大驱动突变 [19];
突变位点:外显子 11 突变占 60%(多见于近端胃 GIST),外显子 9 突变占 6%-8%(多见于小肠 GIST),外显子 13/17 突变多为伊马替尼耐药突变 [19];
3.分子机制:KIT 突变后,无需配体(SCF)即可发生二聚化,激活下游 PI3K/AKT、MAPK、JAK/STAT 通路,促进细胞增殖与抗凋亡 [19];
4.靶向药物研发及临床数据:
伊马替尼(Imatinib)
机制:Ⅰ 型 TKI,抑制 KIT、PDGFRA、ABL 激酶活性 [19];
临床试验:IRIS(Ⅲ 期,晚期 GIST)、SSG-XVIII/AIO(Ⅲ 期,辅助治疗)[19];
临床数据:
——晚期 GIST:KIT 外显子 11 突变患者 ORR 90%,mPFS 18-24 个月;KIT 外显子 9 突变患者需高剂量(800mg/d),ORR 60%[19];
——辅助治疗:高风险 GIST 术后辅助治疗 3 年,无复发生存率(RFS)65.6%,显著优于观察组(47.9%)[19];
——耐药机制:40%-50% 患者因 KIT 外显子 13/17 二次突变耐药 [19];
获批情况:2002 年 FDA 批准用于晚期 GIST,是 GIST 靶向治疗的 “基石”[19];
舒尼替尼(Sunitinib)
机制:Ⅱ 型 TKI,抑制 KIT、PDGFRA/B、VEGFR1-3 [19];
适用场景:伊马替尼耐药 / 不耐受的晚期 GIST [19];
临床试验:EORTC 62024(Ⅲ 期)[19];
临床数据:mPFS 6.8 个月,mOS 26.6 个月,显著优于安慰剂(PFS 1.5 个月,OS 6.5 个月)[19];
优势:对 KIT 外显子 13 突变(如 V654A)敏感,水肿、乏力等不良反应发生率低于伊马替尼 [19];
获批情况:2006 年 FDA 批准用于 GIST 二线治疗 [19];
瑞戈非尼(Regorafenib)
机制:多激酶抑制剂,抑制 KIT、PDGFRA、VEGFR1-3 [19];
适用场景:伊马替尼、舒尼替尼耐药的晚期 GIST [19];
临床试验:GRID(Ⅲ 期)[19];
临床数据:mPFS 4.8 个月,显著优于安慰剂(0.9 个月)[19];
获批情况:2013 年 FDA 批准用于 GIST 三线治疗 [19]。
二
PDGFRA
1. 生物学意义:血小板衍生生长因子受体 α,与 KIT 同属 Ⅲ 型酪氨酸激酶家族,突变后驱动 GIST 发生,尤其与野生型 GIST 相关 [19];
2. 异常特征:
突变率:约 5%-10%[19];
突变位点:以激酶结构域 D842V 突变为主(占 PDGFRA 突变的 70%),该突变对伊马替尼原发耐药 [19];
3.分子机制:PDGFRA 突变后,自发形成二聚体激活下游信号,与 KIT 突变共享 PI3K/AKT、MAPK 通路,促进细胞增殖 [19];
4.靶向药物研发及临床数据:
阿伐替尼(Avapritinib)
机制:Ⅰ 型 TKI,选择性抑制 PDGFRA D842V 突变,对野生型 PDGFRA 抑制活性低 [19];
临床试验:NAVIGATOR(Ⅰ/Ⅱ 期)[19];
临床数据:PDGFRA D842V 突变患者中,ORR 86%,mPFS 24.0 个月,是首个对该突变有效的药物 [19];
获批情况:2020 年 FDA 批准用于 PDGFRA D842V 突变 GIST [19];
瑞派替尼(Ripretinib)
机制:广谱 KIT/PDGFRA 抑制剂,通过 “双重开关控制” 抑制激活环突变(包括 PDGFRA D842V)[19];
临床数据:PDGFRA D842V 突变患者中,ORR 73%,mPFS 12.2 个月 [19];
获批情况:2020 年 FDA 批准用于 GIST 四线治疗,可覆盖 PDGFRA 耐药突变 [19]。
三
SDH 家族缺陷(SDHA/B/C/D 突变)
1.生物学意义:
SDH(琥珀酸脱氢酶)是三羧酸循环及氧化磷酸化的关键酶,缺陷后导致代谢紊乱,驱动野生型 GIST(无 KIT/PDGFRA 突变)发生 [19];
2.异常特征:
发生率:占野生型 GIST 的 50%,由 SDHA/B/C/D 基因突变导致 [19];
临床特征:多发生于年轻患者,常伴胃肠道外转移(如淋巴结、肺)[19];
3.分子机制:
SDH 缺陷导致琥珀酸堆积,抑制 α- 酮戊二酸依赖的双加氧酶,导致 HIF-1α 稳定表达,促进血管生成与肿瘤增殖 [19];
4.靶向药物研发及临床数据:
替莫唑胺(Temozolomide)
机制:烷化剂,通过 DNA 甲基化发挥抗肿瘤作用,SDH 缺陷 GIST 对其敏感 [19];
临床数据:回顾性研究显示,SDH 缺陷 GIST 患者接受替莫唑胺治疗,ORR 35%,mPFS 8.5 个月 [19];
抗血管生成药物(如舒尼替尼)
临床数据:mPFS 6.2 个月,适用于替莫唑胺耐药患者 [19]。
四
NF1 突变
1.生物学意义:NF1(神经纤维瘤病 1 型)是 RAS-GAP 蛋白,正常情况下促进 RAS-GTP 水解为 RAS-GDP,抑制 RAS 信号;突变后失活,导致 RAS-MAPK 通路持续激活 [1,19];
2.异常特征:
发生率:占野生型 GIST 的 10%-15%[1,19];
临床特征:常伴神经纤维瘤病体征,肿瘤多为多发 [1,19];
3.分子机制:NF1 突变后,RAS-GAP 功能丧失,RAS 持续激活 MAPK 通路,驱动肿瘤增殖 [1,19];
4.靶向药物研发及临床数据:
多激酶抑制剂(如瑞派替尼、索拉非尼)
机制:抑制 RAS 下游 MAPK 通路(RAF/MEK/ERK)及其他激酶 [1,19];
临床数据:瑞派替尼治疗 NF1 突变 GIST,ORR 28%,mPFS 7.3 个月 [1,19];
MEK 抑制剂(如曲美替尼)
临床数据:联合多激酶抑制剂,mPFS 提升至 9.1 个月,处于 Ⅱ 期试验 [1,19]
肝细胞癌(HCC)特异性靶点
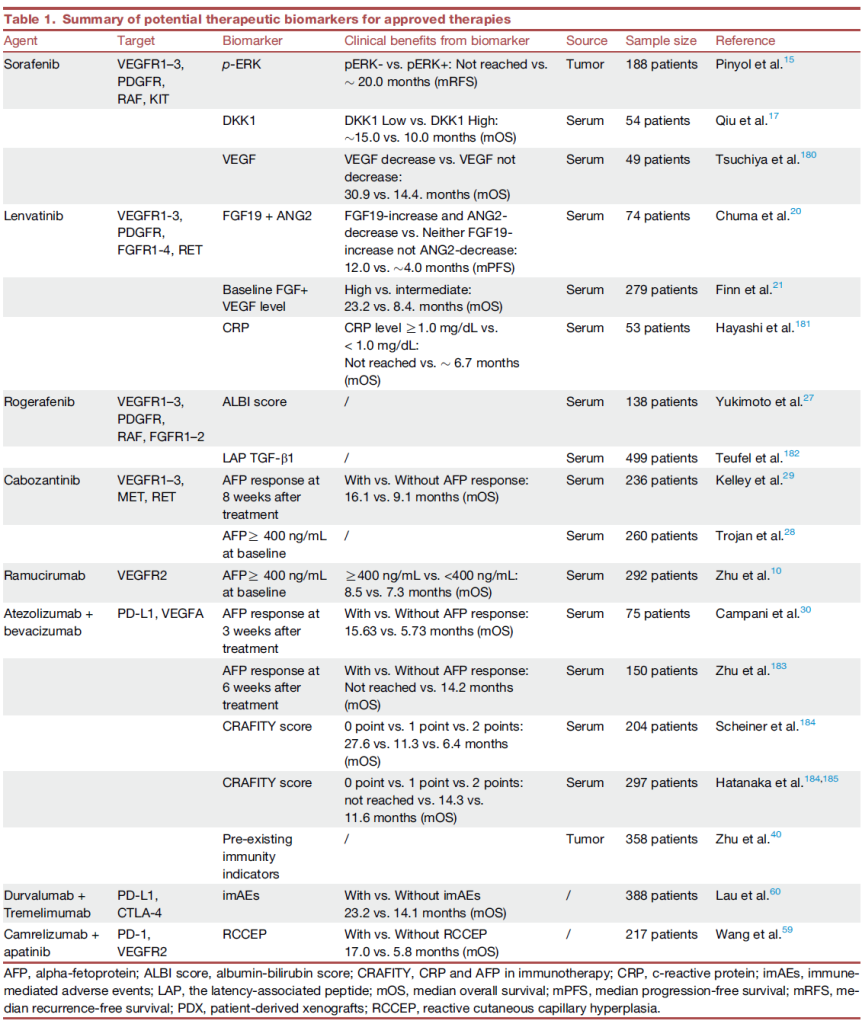
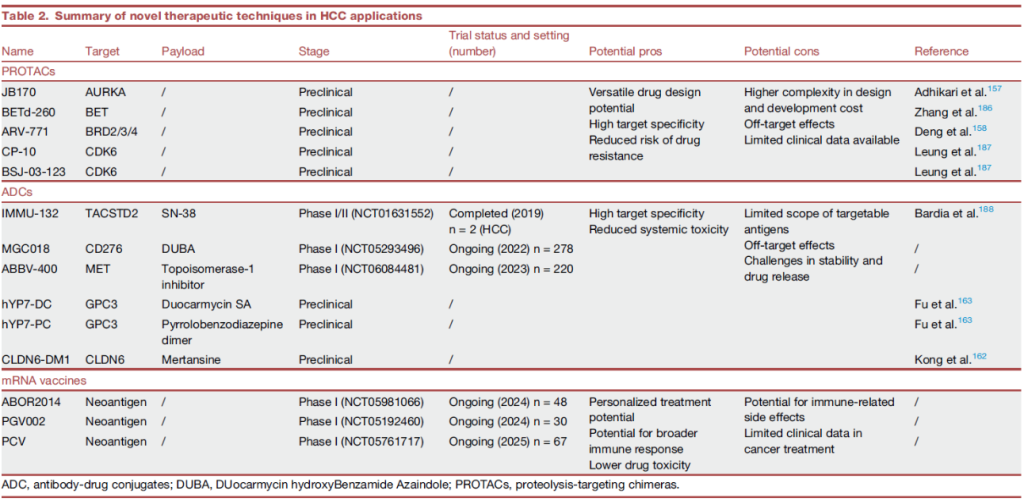
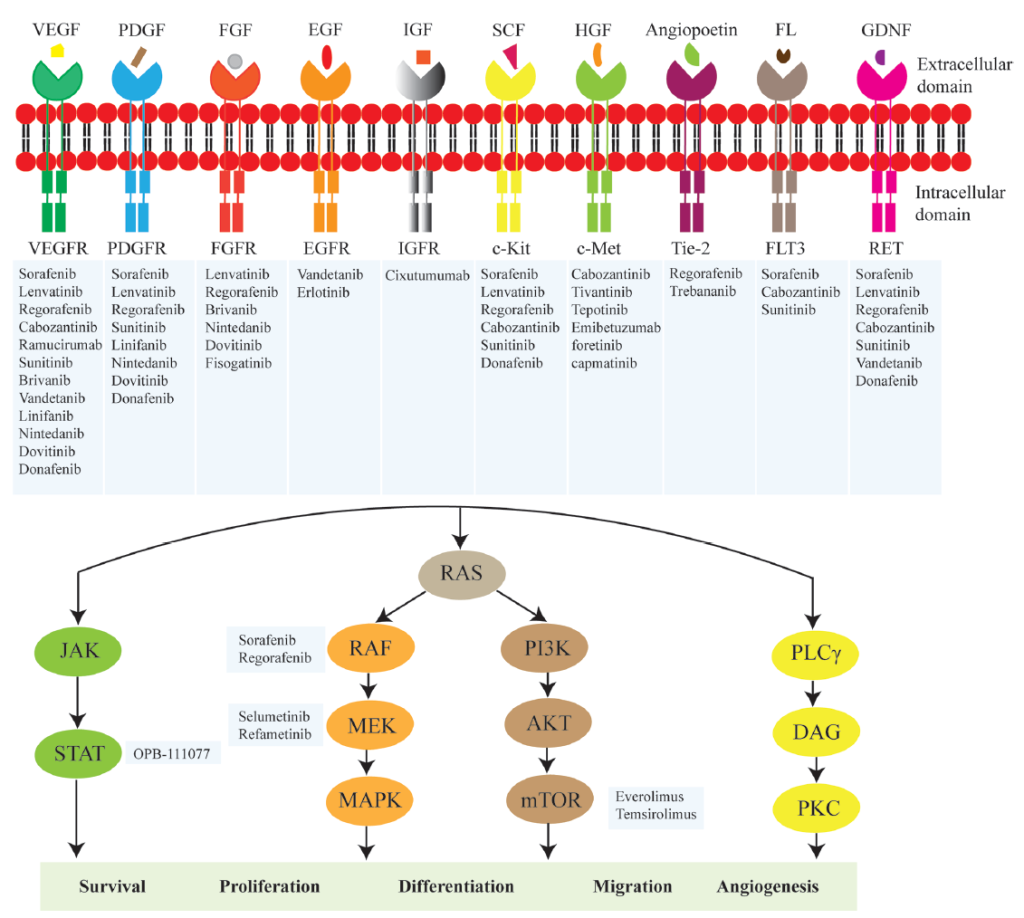
Main molecules of targeted therapy for hepatocellular carcinoma (HCC).
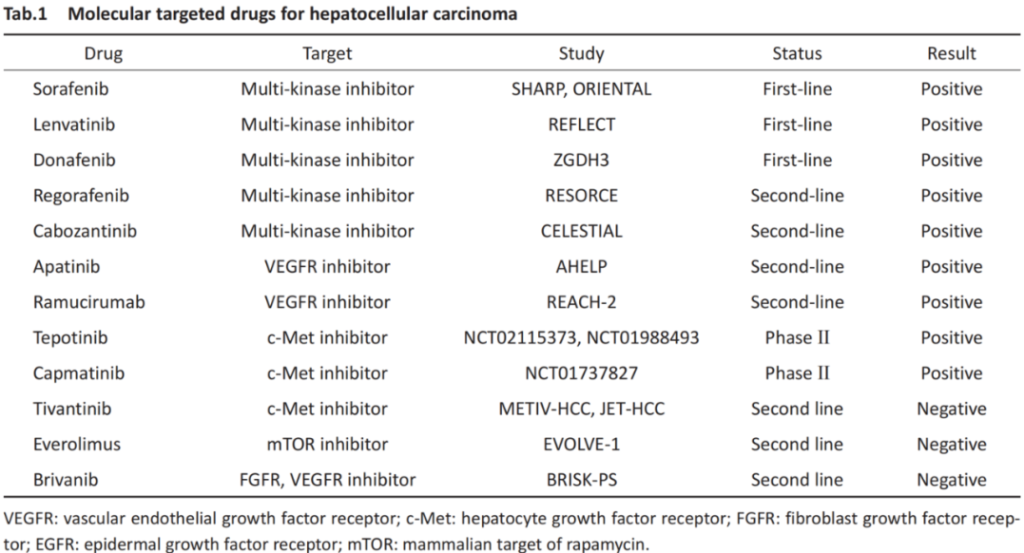
一
MET
1. 生物学意义:MET 是肝细胞生长因子(HGF)受体,调控肝细胞增殖与修复;扩增后激活下游信号,促进肝癌侵袭、转移及耐药 [14,17,18];
2. 异常特征:
扩增率:约 5%-10%[14,17,18];
临床关联:与肝癌血管侵犯、术后复发及索拉非尼 / 仑伐替尼耐药相关 [14,17,18];
3.分子机制:
MET 扩增后,与 HGF 结合或自发二聚化,激活 PI3K/AKT/mTOR、MAPK 通路,促进 EMT 与肿瘤干细胞存活 [14,17,18];
4.靶向药物研发及临床数据:
卡博替尼(Cabozantinib)
机制:多激酶抑制剂,抑制 MET、VEGFR2、AXL 等 [14,17,18];
适用场景:索拉非尼 / 仑伐替尼耐药的晚期肝癌 [14,17,18];
临床试验:CELESTIAL(Ⅲ 期)[14,17,18];
临床数据:二线治疗 mOS 10.2 个月,AFP 高表达患者仍可获益(mOS 9.1 个月)[14,17,18];
特泊替尼(Tepotinib)
机制:高选择性 MET 抑制剂 [14,17,18];
临床试验:Ⅰ/Ⅱ 期试验(NCT03929011)[14,17,18];
临床数据:MET 扩增肝癌患者 ORR 22%,mPFS 5.4 个月 [14,17,18];
埃万妥单抗(Amivantamab,MET/EGFR 双抗):
机制:同时抑制 MET 与 EGFR,阻断双通路激活 [14,17,18];
临床数据:Ⅰ 期试验中,MET 扩增耐药肝癌 ORR 25%,mPFS 6.1 个月 [14,17,18]。
二
AXL
1. 生物学意义:AXL 是 TAM 家族受体酪氨酸激酶,调控细胞存活、EMT 及免疫逃逸,高表达与肝癌不良预后相关 [14,17,18];
2. 异常特征:
高表达率:约 30%[14,17,18];
临床关联:是肝癌血管侵犯、索拉非尼耐药的标志物 [14,17,18];
3.分子机制:
AXL 与配体 GAS6 结合后,激活 PI3K/AKT、MAPK 通路,促进 EMT;同时上调 PD-L1 表达,增强免疫抑制 [14,17,18];
4.靶向药物研发及临床数据:
Bemcentinib(BGB324)
机制:选择性 AXL 抑制剂 [14,17,18];
临床试验:Ⅰ/Ⅱ 期试验(联合索拉非尼)[14,17,18];
临床数据:联合治疗组 mPFS 6.9 个月,显著优于索拉非尼单药(3.7 个月)[14,17,18];
卡博替尼(Cabozantinib)
临床数据:AXL 高表达肝癌患者接受卡博替尼治疗,mOS 11.3 个月,优于 AXL 低表达患者(8.2 个月)[14,17,18]。
三
RET 融合
1. 生物学意义:RET 是受体酪氨酸激酶,融合后持续激活下游信号,驱动肝癌增殖 [14,17,18];
2. 异常特征:
发生率:约 1%-2%[14,17,18];
融合伴侣:常见 CCDC6-RET、NCOA4-RET [14,17,18];
3.分子机制:RET 融合蛋白无需配体即可激活 PI3K/AKT、MAPK 通路,促进细胞增殖 [14,17,18];
4.靶向药物研发及临床数据:
塞尔帕替尼(Selpercatinib)
机制:高选择性 RET 抑制剂 [14,17,18];
临床试验:LIBRETTO-001(Ⅰ/Ⅱ 期,泛癌种 RET 融合)[14,17,18];
临床数据:肝癌 RET 融合患者 ORR 20%,mPFS 4.8 个月 [14,17,18];
普拉替尼(Pralsetinib)
临床数据:RET 融合肝癌 ORR 18%,mPFS 5.1 个月 [14,17,18]。
四
PI3K/AKT/mTOR 通路异常(PI3KCA 突变、PTEN 失活)
1. 生物学意义:PI3K/AKT/mTOR 通路调控细胞代谢、增殖与凋亡,异常激活驱动肝癌代谢重编程(如沃伯格效应)[14,17,18];
2. 异常特征:
异常率:约 30%-50% 肝癌存在 PI3KCA 突变或 PTEN 失活 [14,17,18];
临床关联:与肝癌分化差、转移潜能高相关 [14,17,18];
3.分子机制:
PI3KCA 突变:激活 PI3K 催化亚基,促进 PIP2 转化为 PIP3;
PTEN 失活:无法降解 PIP3,导致 AKT 持续激活,进而激活 mTOR,促进蛋白质合成与代谢 [14,17,18];
4.靶向药物研发及临床数据:
mTOR 抑制剂(依维莫司,Everolimus)
机制:抑制 mTORC1 活性 [14,17,18];
临床试验:RAD001 HCC(Ⅱ 期)[14,17,18];
临床数据:二线治疗 mPFS 3.0 个月,OS 8.5 个月,适用于 PI3K/AKT 激活患者 [14,17,18];
PI3K 抑制剂(阿培利司,Alpelisib)
机制:选择性 PI3Kα 抑制剂 [14,17,18];
临床试验:Ⅰ/Ⅱ 期试验(联合仑伐替尼)[14,17,18];
临床数据:联合治疗组 mPFS 7.2 个月,ORR 28%[14,17,18]。
五
肝癌治疗相关生物标志物(疗效预测靶点)
1. 甲胎蛋白(AFP):
生物学意义:肝癌特异性标志物,与肿瘤增殖活性相关 [14,17,18];
临床应用:预测卡博替尼疗效:AFP≥400ng/mL 患者接受卡博替尼治疗,mOS 9.1 个月;预测雷莫芦单抗疗效:AFP≥400ng/mL 患者二线治疗 mOS 8.5 个月;预测免疫联合疗效:阿替利珠单抗 + 贝伐珠单抗治疗中,AFP 响应(治疗 8 周下降≥50%)患者 mOS 15.63 个月,无响应者 5.73 个月 [14,17,18]。
2. p-ERK(MAPK 通路激活标志物):
临床应用:预测索拉非尼敏感性,p-ERK 阳性患者 mRFS 约 20.0 个月,阴性患者未达到 [14,17,18];
3.血清 FGF19、VEGF:
临床应用:与仑伐替尼响应相关,FGF19 高表达且 VEGF 下降患者 mPFS 12.0 个月,显著优于其他患者(4.0 个月)[14,17,18];
4.CRAFITY 评分(CRP+AFP):
计算方式:CRP≥1.0mg/dL 计 1 分,AFP≥400ng/mL 计 1 分,共 0-2 分 [14,17,18];
临床应用:预测免疫联合疗效,0 分患者 mOS 27.6 个月,1 分 11.3 个月,2 分 6.4 个月 [14,17,18]。
胆道肿瘤(胆管癌、胆囊癌)特异性靶点
一
FGFR2 融合
1. 生物学意义:FGFR2(成纤维细胞生长因子受体 2)融合后持续激活下游信号,促进胆管癌增殖与血管生成,是肝内胆管癌的关键靶点 [7];
2. 异常特征:
发生率:约 10%-15%,肝内胆管癌发生率高于肝外胆管癌 [7];
融合伴侣:常见 BICC1-FGFR2、CCDC6-FGFR2 [7];
3.分子机制:
FGFR2 融合蛋白无需配体即可激活 PI3K/AKT、MAPK 通路,促进细胞增殖与血管生成 [7];
4.靶向药物研发及临床数据:
培米替尼(Pemigatinib)
机制:选择性 FGFR1-3 抑制剂 [7];
临床试验:FIGHT-202(Ⅱ 期)[7];
临床数据:FGFR2 融合胆管癌患者 ORR 36%,mPFS 6.9 个月,mOS 21.1 个月 [7];
获批情况:2020 年 FDA 批准用于 FGFR2 融合胆管癌 [7];
英菲格拉替尼(Infigratinib)
机制:FGFR1-3 抑制剂 [7];
临床试验:PROOF(Ⅱ 期)[7];
临床数据:ORR 23%,mPFS 7.3 个月 [7];
获批情况:2021 年 FDA 批准用于 FGFR2 融合胆管癌 [7]。
二
IDH1 突变
1. 生物学意义:IDH1(异柠檬酸脱氢酶 1)正常情况下催化异柠檬酸转化为 α- 酮戊二酸;突变后催化生成 2 – 羟基戊二酸(2-HG),抑制表观遗传调控酶,驱动胆管癌发生 [7];
2. 异常特征:
发生率:约 10%-20%,以 R132H 突变为主(占 IDH1 突变的 80%)[7];
临床关联:多见于肝内胆管癌,与年轻患者、低分化肿瘤相关 [7];
3.分子机制:
2-HG 堆积抑制 α- 酮戊二酸依赖的双加氧酶(如组蛋白去甲基化酶、TET 甲基胞嘧啶双加氧酶),导致组蛋白修饰异常及 DNA 高甲基化,沉默肿瘤抑制基因 [7];
4.靶向药物研发及临床数据:
艾伏尼布(Ivosidenib)
机制:选择性 IDH1 R132H 抑制剂,降低 2-HG 水平 [7];
临床试验:ClarIDHy(Ⅲ 期)[7];
临床数据:IDH1 突变胆管癌患者,艾伏尼布组 mPFS 2.7 个月,显著优于安慰剂组(1.4 个月);OS 有延长趋势(10.3 个月 vs 7.5 个月)[7];
获批情况:2021 年 FDA 批准用于 IDH1 突变胆管癌 [7];
恩西地平(Enasidenib,IDH2 抑制剂)
临床数据:IDH2 突变胆管癌患者 ORR 15%,mPFS 4.8 个月,处于 Ⅱ 期试验 [7]。
三
KRAS 扩增
1. 生物学意义:KRAS 扩增后激活下游 MAPK/PI3K 通路,促进胆管癌增殖与免疫逃逸 [7];
2. 异常特征:
发生率:约 15%-20%[7];
临床关联:与 PD-L1 表达负相关,患者对单纯免疫治疗响应差 [7];
3. 分子机制:KRAS 扩增导致蛋白过表达,持续激活 MAPK 通路,同时上调免疫抑制因子(如 IL-10),抑制 CD8+T 细胞浸润 [7];
4. 靶向药物研发及临床数据:
KRAS G12C 抑制剂(索托拉西布、阿达格拉西布)
适用人群:KRAS G12C 突变胆管癌(发生率约 2%-3%)[7];
临床数据:Ⅰ/Ⅱ 期试验显示 ORR 15%-20%,mPFS 4.0-5.2 个月 [7];
联合方案(KRAS 抑制剂 + PD-1 抑制剂)
机制:KRAS 抑制剂降低免疫抑制,增强 PD-1 抑制剂疗效 [7];
临床数据:Ⅰ 期试验显示 ORR 28%,mPFS 6.5 个月,处于 Ⅱ 期验证阶段 [7]。
四
BRAF V600E 突变
1. 生物学意义:BRAF V600E 突变持续激活 MAPK 通路,促进胆管癌增殖 [7];
2. 异常特征:
发生率:约 5%-7%[7];
临床关联:与肿瘤高侵袭性、不良预后相关 [7];
3.分子机制:同 “泛消化道肿瘤共性靶点 – BRAF 靶点”,突变后独立激活 MEK/ERK 信号 [7];
4.靶向药物研发及临床数据:
达拉非尼(Dabrafenib)+ 曲美替尼(Trametinib):
机制:BRAF 抑制剂 + MEK 抑制剂,双重阻断 MAPK 通路 [7];
临床试验:Ⅰ/Ⅱ 期试验 [7];
临床数据:BRAF V600E 突变胆管癌 ORR 51%,mPFS 9.1 个月,mOS 14.2 个月 [7];
恩科拉非尼(Encorafenib)+ 比尼美替尼(Binimetinib)
临床数据:ORR 47%,mPFS 8.3 个月 [7]。
消化道肿瘤精准治疗的未来展望
尽管消化道肿瘤靶向治疗已取得显著进展,但仍面临 “靶点覆盖不足、耐药频发、疗效异质性” 等挑战[1,2,3]。未来需从 “机制探索、药物研发、技术应用、临床转化” 四方面突破,推动精准治疗向 “更广泛、更深入、更个体化” 发展[2,3,6,7,14,15,16]。
一
联合治疗:从 “单靶点” 到 “多通路协同”
1. 靶点联合策略
KRAS 抑制剂联合 EGFR 抑制剂:如索托拉西布 + 西妥昔单抗,通过 “抑制 KRAS 下游 + 阻断 EGFR 反馈激活”,克服 KRAS 抑制剂单药耐药,目前 CodeBreaK300Ⅲ 期试验已证实该方案在结直肠癌中的优势[6,7,15];
BRAF+MEK+EGFR 联合:如恩科拉非尼 + 比尼美替尼 + 西妥昔单抗,三重阻断 MAPK 通路,避免 “通路旁路激活”,BEACON CRC 试验已证实其在 BRAF V600E 突变结直肠癌中的疗效[15,16];
肝癌特异性联合策略:KRAS 抑制剂(如 BI 1701963)联合 MET 抑制剂,针对肝癌 KRAS-MET 共激活患者,临床前模型中位 PFS 延长至 8.3 个月 [14]。
2.抗血管生成 + 免疫治疗:如贝伐珠单抗 + 阿替利珠单抗,通过 “血管正常化 + 解除免疫抑制”,增强 T 细胞浸润,已成为肝癌、结直肠癌的一线方案[15,17,18];
3.表观遗传药物联合:HDAC 抑制剂与 DNA 甲基转移酶抑制剂联合,协同逆转肿瘤抑制基因沉默[1]。
二
新型靶点与药物研发:覆盖 “未被满足的需求”
1. 泛 KRAS 抑制剂研发
目前 KRAS G12C 抑制剂仅覆盖 1%-3% 的结直肠癌、13% 的非小细胞肺癌,需开发针对 G12D/V、G13D 等高频突变的泛 KRAS 抑制剂[5,6,7]。
如 BI 1701963(SOS1 抑制剂),通过抑制 KRAS 上游激活因子,间接抑制所有 KRAS 突变亚型,联合 KRAS G12C 抑制剂可延缓耐药 [5,6,7];
新型共价抑制剂:如针对 KRAS G12D 的不可逆抑制剂,通过结合 G12D 突变特有的口袋,已在临床前模型中显示出活性 [5,6,7]。
2.罕见靶点药物开发
NTRK 融合:拉罗替尼(Larotrectinib)、恩曲替尼(Entrectinib)已获批用于泛癌种 NTRK 融合,在结直肠癌、胰腺癌中 ORR 超 70%,需扩大筛查覆盖 [15,16];
RET 融合:塞尔帕替尼(Selpercatinib)在 RET 融合胰腺癌中 ORR 20%,虽低于其他瘤种,但为 RET 阳性患者提供了新选择 [11,14];
FGFR2 融合:培米替尼(Pemigatinib)已获批用于胆管癌,在胃癌、胰腺癌中处于 Ⅰ/Ⅱ 期试验,ORR 约 30% [12,13];
CLCA1、MUC5AC:针对结直肠癌新兴靶点的调节剂在研,有望成为耐药后治疗选择[1];
针对 WNT-β 连环蛋白通路异常,开发 β 连环蛋白降解剂,临床前试验在肝癌模型中肿瘤缩小率超 60%[14]。
3.ADC 药物拓展
针对 HER2、CLDN18.2 的 ADC 药物:如 DS-8201(HER2)、Zolbetuximab-deruxtecan(CLDN18.2),通过优化 linker 和细胞毒性药物,提升疗效并降低毒性 [12,13];
双靶点 ADC:如同时靶向 HER2 和 CLDN18.2 的 ADC,覆盖 HER2 阳性 / CLDN18.2 高表达重叠人群,目前处于临床前研发阶段[12,13]。
三
技术驱动:精准检测与动态监测
1.NGS 全景测序的普及:明确指出,NGS 应作为消化道肿瘤常规检测手段[1,9,14,15,16]。
覆盖靶点:同时检测 KRAS、NRAS、BRAF、HER2、NTRK、RET 等靶点,避免 “单一靶点检测遗漏”[9,15,16];
液体活检:循环肿瘤 DNA(ctDNA)检测可动态监测治疗反应,如在结直肠癌中,ctDNA 检测 KRAS 突变可提前 10 个月预测抗 EGFR 治疗耐药,指导方案调整[1,15,16];
肝癌非侵入性检测:液体活检(ctDNA)检测肝癌 KRAS、MET 突变,可提前 10 个月预测索拉非尼 / 仑伐替尼耐药,指导方案调整;AI 影像分析(如 MRI 结合深度学习)可无创评估肝癌 TME 炎症状态,准确率约 85%[14]。
2.空间多组学技术应用
空间转录组学:如 GeoMX、CODEX 技术,可在肿瘤组织空间维度上解析免疫细胞浸润、信号通路激活状态,指导 “个体化联合方案”(如肿瘤微环境 “冷” 区域需联合抗血管生成药物,“热” 区域需联合 ICIs) [1,14];
单细胞测序:解析肿瘤内异质性,如结直肠癌中 KRAS 突变亚克隆与野生型亚克隆的共存情况,指导 “联合靶点选择”[1,14]。
3.类器官研究应用:患者来源类器官(PDOs)可复刻肝癌肿瘤特征,用于体外药物筛选,筛选准确率约 80%;异种移植模型(PDXs)评估新型 ADC 药物疗效,与临床响应一致性达 75%[14]。
四
克服耐药:从 “被动应对” 到 “主动预防”
1.耐药机制解析
基因组层面:通过 ctDNA 动态监测耐药突变,如 KRAS 抑制剂耐药的 EGFR S492R、MET 扩增,及时调整联合方案[6,7,14,15];
表观遗传层面:如 DNA 甲基化导致 MLH1 沉默,可联合 DNA 甲基转移酶抑制剂(如 5 – 氮杂胞苷),恢复 MLH1 表达,增强 ICIs 疗效 [1,2,14];
通路代偿层面:Notch、Hippo 通路代偿激活导致的耐药,可联合对应通路抑制剂[1]。
2.适应性治疗策略
基于 ctDNA 的 “剂量调整”:如根据 ctDNA 中肿瘤突变 allele 分数(AF)调整药物剂量,避免 “过度治疗” 或 “治疗不足” [8,14];
轮换治疗:如 KRAS 抑制剂与 EGFR 抑制剂轮换使用,避免单一靶点耐药克隆扩增,目前在临床前模型中已显示出延长 PFS 的效果 [6,7,15]。
五
公平可及与质量控制:推动精准治疗落地
1.标准化检测流程
CLDN18.2 IHC 检测:明确标本固定时间(活检标本 6-8 小时,手术标本 24-48 小时)、抗体选择(如 43-14A、14G11)及判读标准(IHC 2+/3+≥75% 为高表达),避免 “假阴性”[12,13];
MSI-H/dMMR 检测:推荐 “IHC+PCR” 联合检测,互补优缺点,提高准确率 [4,15,16];
表观遗传标志物检测:标准化 DNA 甲基化、组蛋白修饰检测流程,确保结果一致性[1]。
2.扩大临床研究覆盖
纳入罕见亚型:如早发性结直肠癌(EOCRC)、KRAS 野生型胰腺癌,这些亚型具有独特分子特征,需针对性开展临床试验 [11,15];
覆盖不同地区人群:如亚洲人群与欧美人群的 HER2、CLDN18.2 表达率存在差异,需开展多中心研究,验证药物在不同人群中的疗效 [12,13]。
总 结
消化道肿瘤精准治疗已从 “探索阶段” 进入 “成熟应用阶段”:KRAS、EGFR/HER2、BRAF 等核心靶点的分子机制已明确,对应的药物(如索托拉西布、西妥昔单抗、恩科拉非尼)已通过临床试验验证,成为标准治疗方案。新增的 Notch、Hippo、组蛋白修饰等通路进一步丰富了治疗靶点库,为耐药患者提供了新方向。
未来需进一步突破 “泛靶点药物研发、耐药机制克服、技术精准应用” 三大方向,通过 “多学科协作(MDT)” 整合基因组学、影像学、临床数据,为患者制定 “量体裁衣” 的治疗方案,最终实现 “从癌症治疗到癌症管理” 的转变,改善全球消化道肿瘤患者的预后。
参考文献
1.Qing Li, et al. Signaling pathways involved in colorectal cancer: pathogenesis and targeted therapy. Signal Transduction and Targeted Therapy (2024) 9:266
2.Leowattana W, Leowattana P, Leowattana T. Systemic treatment for metastatic colorectal cancer. World J Gastroenterol. 2023 Mar 14;29(10):1569-1588.
3.Underwood PW, Ruff SM, Pawlik TM. Update on Targeted Therapy and Immunotherapy for Metastatic Colorectal Cancer. Cells. 2024 Jan 28;13(3):245.
4.Ludford K, Ho WJ, Thomas JV, et al. Neoadjuvant pembrolizumab in localized microsatellite instability high/deficient mismatch repair solid tumors. J Clin Oncol, 2023, 41(12): 2181-2190.
5.Johnson C, et al. Classification of KRAS-Activating Mutations and the Implications for Therapeutic Intervention. Cancer Discov,2022,12(4):913-923.
6.Tanaka N, et al. Mechanisms of Resistance to KRAS Inhibitors: Cancer Cells’Strategic Use of Normal Cellular Mechanisms to Adapt. Cancer Sci,2025,116(3):600-612.
7.Shi Y, et al .Targeting KRAS: From Metabolic Regulation to Cancer Treatment.Mol Cancer,2025,24(1):9.
8.Kuang J, et al. Genomic and microenvironmental insights into drug resistance in colorectal cancer liver metastases. Discov Oncol,2025,16(1):241.
9.Wu L, et al. Pan-cancer analysis to characterize the clinicopathological and genomic features of KRAS-mutated patients in China. J Cancer Res Clin Oncol,2025,151(2):94.
10.Ash LJ, et al. KRAS: Biology, Inhibition, and Mechanisms of Inhibitor Resistance. Curr Oncol,2024,31(4):2024-2046.
11.Jin D, Khan NU, Gu W, Lei H, Goel A, Chen T. Informatics strategies for early detection and risk mitigation in pancreatic cancer patients. Neoplasia. 2025 Feb;60:101129.
12.Dong Luo, Yunmei Liu, Zhengmao Lu and Lei Huang. Targeted therapy and immunotherapy for gastric cancer: rational strategies, novel advancements, challenges, and future perspectives. Molecular Medicine (2025) 31:52
13.Raghav Sundar, et al. Gastric cancer. The Lancet. May 1, 2025.
14.Xupeng Yang, et al. Precision treatment in advanced hepatocellular carcinoma. Cancer Cell 42, February 12, 2024.
15.Xie YH, Chen YX, Fang JY. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct Target Ther. 2020 Mar 20;5(1):22.
16.Simona Gabriela Duta-Ion, et al. Redefining Therapeutic Approaches in Colorectal Cancer: Targeting Molecular Pathways and Overcoming Resistance. Int. J. Mol. Sci. 2024, 25, 12507.
17.Niu M, Yi M, Li N, Wu K, Wu K. Advances of Targeted Therapy for Hepatocellular Carcinoma. Front Oncol. 2021 Jul 26;11:719896.
18.Ao Huang, et al. Targeted therapy for hepatocellular carcinoma. Signal Transduction and Targeted Therapy volume 5, Article number: 146 (2020).
19.Unk, M.; Jezeršek Novakovi´c, B.; Novakovi´c, S. Molecular Mechanisms of Gastrointestinal Stromal Tumors and Their Impact on Systemic Therapy Decision. Cancers 2023, 15, 1498.