


实体瘤中的药物基因组学:遗传变异及其临床应用
- boke
- 2025-12-01
- 5:50 下午
药物基因组学是研究遗传变异如何影响药物反应的学科,随着精准医疗的发展,已成为癌症治疗中不可或缺的部分。肿瘤患者的遗传异质性显著影响药物疗效和毒性,将药物基因组学检测纳入临床实践可以有效指导药物选择和剂量调整并有助于识别发生严重毒性反应风险较高的患者。
细胞色素 P450 酶、DPYD 和 UGT1A1 等关键基因已被确定为影响他莫昔芬、环磷酰胺、酪氨酸激酶抑制剂、氟尿嘧啶类药物和伊立替康等常用化疗药物代谢的重要生物标志物,进而导致患者治疗结局存在差异。
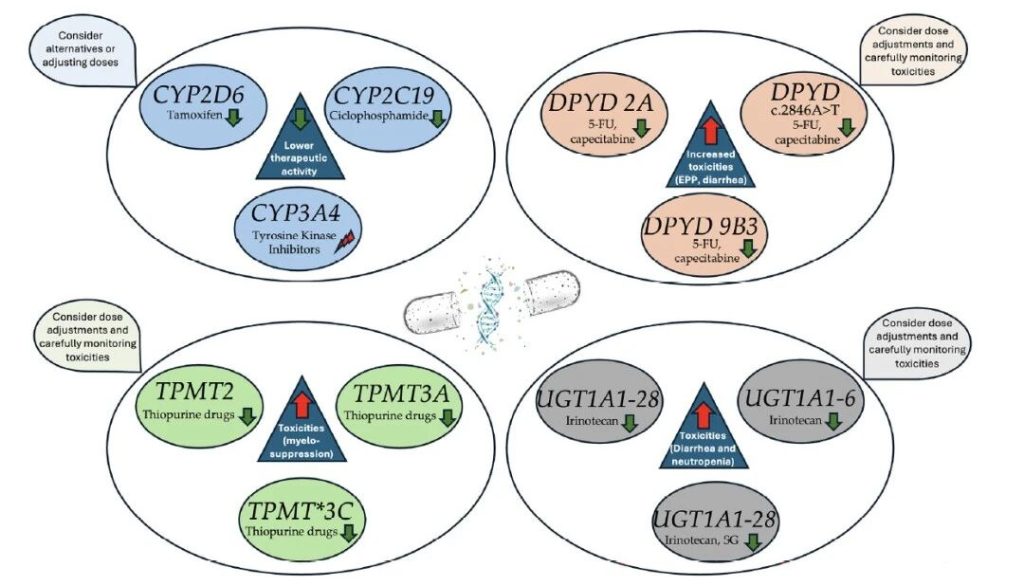
Fig.Key pharmacogenomic genes with clinical implicationsin solid tumors.Green arrows indicate a reduction in function, while red arrows indicate an increase.The beam represents dysfunction which can imply either an increase or a decrease in enzyme function.
一
药物代谢动力学与药物效应动力学相关的基因多态性
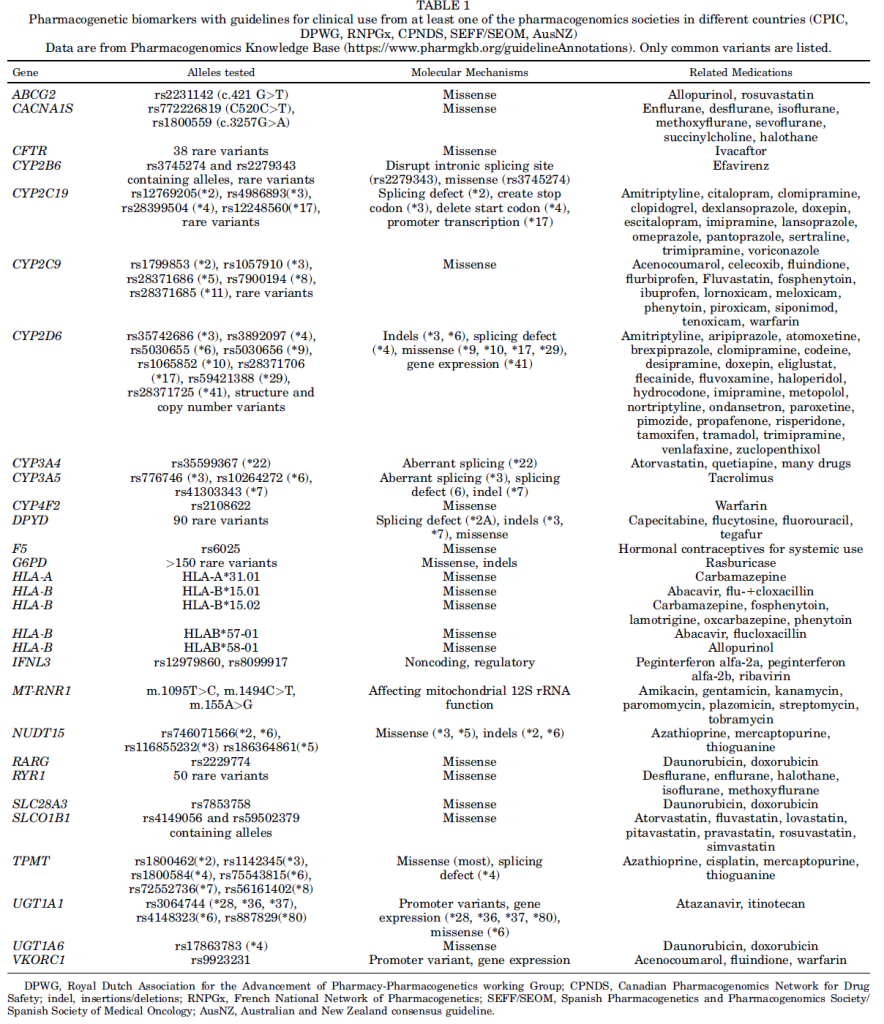
药物代谢酶基因
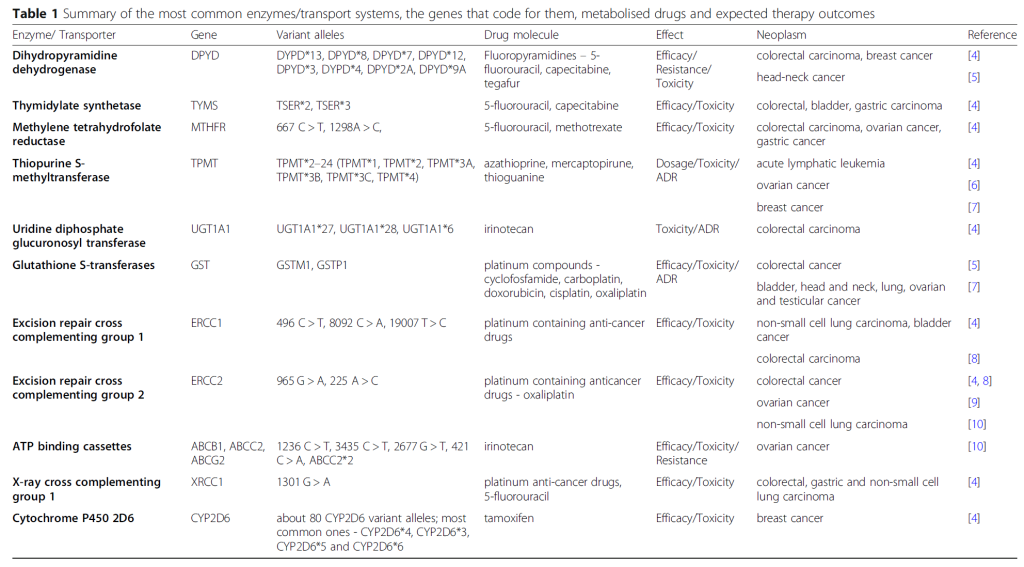
药物代谢酶基因通过调控化疗药物的激活、灭活或解毒过程,直接影响药物的血药浓度和活性代谢产物生成,是药物反应个体差异的主要来源。
1.CYPs(细胞色素 P450 酶)
CYPs 是一个超家族酶,在多种化疗药物代谢中发挥关键作用,其活性由遗传多态性决定,导致个体间药代动力学差异显著。
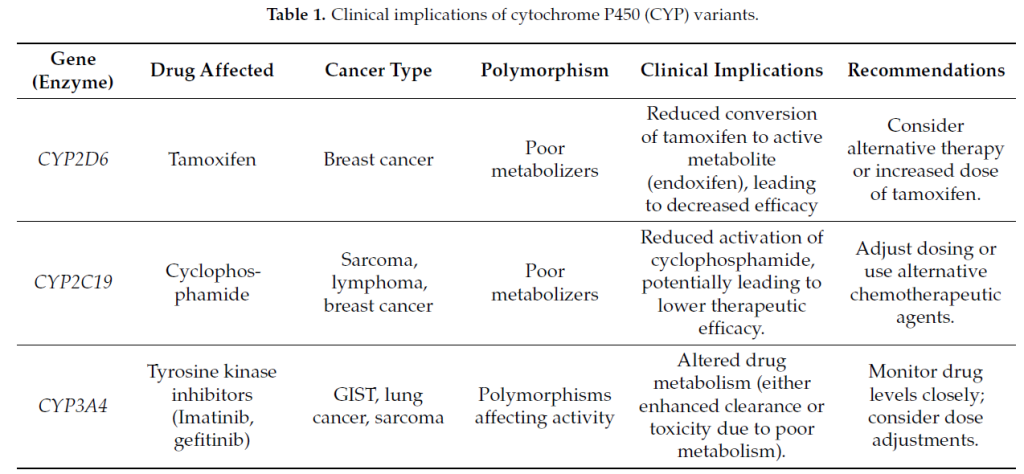
1.1 CYP2D6 与他莫昔芬代谢
他莫昔芬——一种选择性雌激素受体调节剂(SERM),作为前药,需通过 CYP2D6 代谢激活为最有效的活性代谢产物恩多昔芬,该代谢产物对于在乳腺组织中发挥药物的抗雌激素作用、抑制癌细胞增殖和降低复发风险至关重要。
CYP2D6 酶活性的差异会影响他莫昔芬治疗的效果。目前已知有超过 100 种CYP2D6多态性会影响酶活性,导致表型从弱代谢者(PM)到超快代谢者(UM)不等。
弱代谢者的酶活性降低或缺失,导致活性代谢产物恩多昔芬水平降低,可能导致治疗效果不佳。
超快代谢者的 CYP2D6 活性增强,产生更高浓度的恩多昔芬,可能获得更好的治疗反应。
部分临床指南推荐进行 CYP2D6 基因分型,但由于各研究结果不一致,其常规应用仍存在争议 。
1.2 CYP2C19 与环磷酰胺激活
CYP2C19 是细胞色素 P450 家族的另一种重要酶,在包括环磷酰胺在内的多种化疗药物代谢中发挥关键作用。
环磷酰胺是一种前药,常用于治疗淋巴瘤、乳腺癌和卵巢癌,需通过 CYP2C19 和其他酶(如 CYP2B6)进行生物激活,产生对癌细胞具有细胞毒性的活性代谢产物。
CYP2C19 基因的遗传多态性导致不同的代谢者表型。
弱代谢者的酶活性降低,可能会影响环磷酰胺的激活,减少活性代谢产物的产生,从而可能降低治疗效果。
超快代谢者的酶活性增强,可能会提高药物激活效率和疗效,但同时也会增加骨髓抑制等毒性反应的风险。
目前,通过 CYP2C19 基因分型指导环磷酰胺剂量调整的临床应用仍在研究中。初步研究表明,CYP2C19 活性降低的患者可能需要通过剂量调整或替代治疗来优化治疗效果。
1.3 CYP3A4 与酪氨酸激酶抑制剂(TKIs)
CYP3A4 是细胞色素 P450 家族的关键酶,负责代谢约 50% 的临床常用药物,包括伊马替尼、厄洛替尼和吉非替尼等多种酪氨酸激酶抑制剂,这些药物用于治疗慢性粒细胞白血病(CML)和非小细胞肺癌(NSCLC)。
CYP3A4 的遗传变异或环境因素(如同时使用抑制剂或诱导剂)影响药物浓度。
CYP3A4 活性增强可能导致酪氨酸激酶抑制剂清除加快,导致血药浓度低于治疗水平,从而降低治疗效果。
CYP3A4 活性降低可能导致药物浓度升高,增加肝毒性、腹泻和皮疹等不良反应的发生风险 。
鉴于 CYP3A4 活性存在个体差异,虽然目前 CYP3A4 多态性的常规基因检测尚未成为标准做法,但了解个体的代谢能力对于制定个性化治疗方案和加强药物基因组学中的以患者为中心的治疗至关重要。
2.DPYD(二氢嘧啶脱氢酶)
二氢嘧啶脱氢酶(DPYD)基因编码二氢嘧啶脱氢酶(DPD),该酶是氟尿嘧啶类药物(如 5-氟尿嘧啶(5 – FU)、卡培他滨)代谢的关键酶。
2.1 DPYD 的功能及其在氟尿嘧啶类药物代谢中的重要性
5 – FU通过抑制 DNA 合成和修复中的关键酶胸苷酸合酶,阻止肿瘤细胞增殖。只有一小部分 5 – FU转化为氟脱氧尿苷一磷酸(FdUMP)等活性代谢产物,这些活性代谢产物具有细胞毒性。大多数 5 – FU(约 80%)会通过 DPYD 酶快速代谢并失活。
在 DPYD 活性降低或缺失的患者中,未代谢的 5 – FU会在体内蓄积,导致药物暴露量增加,显著提高严重毒性反应的发生风险,包括骨髓抑制(中性粒细胞减少、血小板减少)、胃肠道毒性(腹泻、黏膜炎、恶心)、手足综合征、心脏毒性和神经毒性。
2.2 DPYD 多态性及其临床意义
DPYD 基因具有高度多态性,某些变异会导致酶活性部分或完全缺乏。已有多种特征明确的多态性被证实与 DPYD 功能显著降低相关,每种多态性都对应着不同程度的氟尿嘧啶诱导毒性风险。
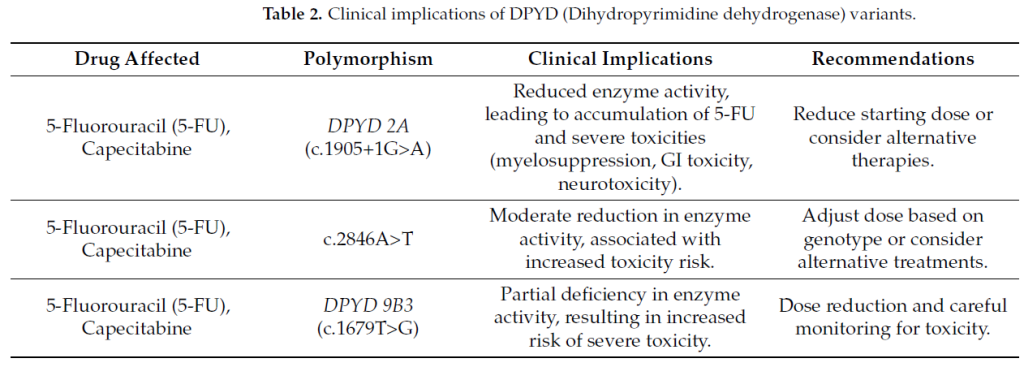
关键的 DPYD 变异包括 :
DPYD2A(c.1905+1G>A),这是临床相关性最强、研究最充分的突变,其通过 mRNA 加工过程中第 14 外显子跳跃,导致酶失去功能,显著增加接受标准剂量氟尿嘧啶类药物治疗患者的严重毒性反应风险。
c.2846A>T(DPYD13)变异影响酶的底物结合位点,与接受 5 – 氟尿嘧啶或卡培他滨治疗患者的中度至重度毒性反应相关。
c.1679T>G(DPYD*9B3)多态性导致氨基酸替换(p.I560S),与 DPYD 活性中度降低相关,增加毒性反应风险。
c.1236G>A(HapB3)变异虽然不直接影响 DPYD 活性,但与功能变异存在连锁不平衡,在部分人群中与毒性反应增加相关。
其他变异:IVS11+1 G>T、731A>C(E244V)、1651 G>A(A551T)、G1601A(DPYD4)等。
由于存在显著风险,包括欧洲肿瘤内科学会(ESMO)和临床药物基因组学实施联盟(CPIC)在内的多个健康组织建议在治疗前对 DPYD 多态性进行基因检测,以识别高风险患者。
DPYD 多态性患者的治疗管理重点在于个性化剂量调整策略。
对于 DPYD 部分缺乏的患者(即 DPYD2A 杂合子携带者),建议将剂量降低 50% 或更多,并进行密切监测。
对于 DPYD 完全缺乏的患者(即 DPYD2A 纯合子携带者),应考虑不涉及 5 – 氟尿嘧啶或卡培他滨的替代化疗方案,以避免危及生命的毒性反应。
治疗药物监测(TDM)和实时 5 – 氟尿嘧啶血浆浓度检测可进一步帮助优化治疗,同时最大限度降低毒性风险。
2.3 研究与未来方向
多态性研究
尽管 DPYD2A、c.2846A>T 和其他常见变异已得到充分研究,但关于可能导致 DPYD 缺乏的其他多态性的研究仍在继续。特别是,DPYD 变异频率的种族差异凸显了制定人群特异性检测方案的必要性。
基因型与表型结合检测
目前正在进行将 DPYD 基因分型与表型检测(如测量血浆中 5 -FU代谢产物二氢尿嘧啶水平)相结合的研究,以更全面地评估 DPYD 功能。这种方法有助于提高识别氟尿嘧啶类药物毒性反应高风险患者的准确性,尤其是对于携带新型或罕见 DPYD 变异的患者。
多基因检测
将 DPYD 检测与其他参与FU类药物代谢的遗传标志物(如胸苷酸合酶(TYMS)和亚甲基四氢叶酸还原酶(MTHFR))相结合,这种多基因检测方法有助于更好地理解个体药物反应和毒性风险,从而制定更精准、个性化的治疗策略。
指南建议
目前,许多临床指南建议在治疗前进行 DPYD 检测,以识别高风险患者并相应地调整化疗方案。尽管 DPYD 检测的常规应用尚未普及,但正在进行的研究支持其在个性化癌症治疗中的重要性,有望改善患者预后并降低治疗相关发病率。
3.TYMS(胸苷酸合成酶)
DPD 并非唯一参与 5-FU 代谢的酶,胸苷酸合成酶也参与该药物的作用通路。TS 与胸苷合成相关,是 5-FU 的作用靶点,5-FU 可通过抑制 TS 发挥作用。
编码 TS 的基因为 TYMS,已发现该基因存在两种不同的等位基因——含 2 个重复序列的 TSER2 和含 3 个重复序列的 TSER3。
结直肠癌患者接受 5-FU 治疗时,携带 TSER3 等位基因的患者结局优于携带 TSER2 多态性的患者。然而,并非所有携带 TSER3 等位基因的患者结局都较差,最可能的解释是存在另一种变异(G→C 单核苷酸多态性),该变异导致 TS 活性降低,与 TSER2 等位基因相当。约 29%-57% 的 TSER*3 等位基因中存在该单核苷酸多态性。
联合 TYMS 和 DPYD 基因分型有助于筛选出对 5-FU 治疗反应更佳且不良反应更少的患者。
4.MTHFR(亚甲基四氢叶酸还原酶)
亚甲基四氢叶酸还原酶是一种参与 5-FU 和甲氨蝶呤(MTX)代谢通路的酶,在叶酸和蛋氨酸代谢以及 DNA 合成与甲基化过程中发挥重要作用。MTHFR 可代谢 5-FU 的底物(5,10 – 亚甲基四氢叶酸),因此该酶功能降低会增强 5-FU 的活性,但会降低对 MTX 的敏感性。
编码 MTHFR 的基因中最常见的多态性是 677 C>T。MTHFR 677 C>T 变异是导致 MTHFR 蛋白热不稳定性的致病变异,该热不稳定蛋白在体外的活性降低 50%,其与甲氨蝶呤(用于化疗和抗炎治疗)等药物存在关联:在接受甲氨蝶呤治疗的儿童急性淋巴细胞白血病患者中,T 等位基因与无事件生存率降低相关,但并非毒性或癫痫发作的风险因素。
部分研究表明,在结直肠癌、乳腺癌等疾病的发病率方面,T 等位基因可能比 C 等位基因具有保护作用。
接受 MTX 治疗的卵巢癌患者的研究显示,TT 纯合子基因型患者中 77% 出现 3-4 级毒性,杂合子患者中为 6%,CC 纯合子基因型患者中为 8%。TT 纯合子基因型患者的同型半胱氨酸水平较高,且与毒性增加相关。
此外,TT 纯合子基因型患者接受 5-FU 治疗时结局更佳。一项针对 43 例接受 5-FU 治疗的结直肠癌患者的临床试验显示,TT 纯合子基因型患者的反应率和总生存率均更优。
5.TPMT(硫嘌呤甲基转移酶)
硫嘌呤甲基转移酶(TPMT)是参与硫嘌呤类药物(如硫唑嘌呤、6 – 巯基嘌呤(6-MP)和硫鸟嘌呤)代谢的关键酶。
TPMT 通过催化硫嘌呤类药物的 S – 甲基化反应,减少其向细胞毒性代谢产物的转化,从而发挥关键作用。
由遗传多态性导致的 TPMT 酶活性差异,会显著影响硫嘌呤类药物治疗的疗效和毒性。

5.1 TPMT 遗传变异与临床意义
特征最明确的 TPMT 多态性包括: TPMT2、TPMT3A 和 TPMT*3C。这些遗传变异导致 TPMT 酶活性降低,使得接受标准剂量硫嘌呤类药物治疗的患者体内毒性硫鸟嘌呤核苷酸(TGNs)蓄积。个体可分为正常代谢者(野生型)、中间代谢者(低功能变异杂合子)或弱代谢者(低功能变异纯合子)。
弱代谢者发生严重骨髓抑制的风险极高,骨髓抑制是一种危及生命的不良反应,其特征是骨髓造血功能受抑。
中间代谢者也容易发生硫嘌呤类药物相关毒性反应,但程度低于弱代谢者。
5.2 临床实践中的 TPMT 检测
为降低严重毒性反应的发生风险,临床指南建议在开始硫嘌呤类药物治疗前进行治疗前 TPMT 基因分型或表型分析。
对于携带两个无功能 TPMT 等位基因的患者(弱代谢者),建议将剂量降低高达 90%。
中间代谢者通常需要适度降低剂量并进行密切监测。
若未进行检测,患者往往需要通过试错法调整剂量,这会增加不良结局的发生风险。
5.3 TPMT 与其他治疗药物
尽管 TPMT 多态性主要与硫嘌呤类药物相关,但研究表明,它们也可能影响其他治疗药物的代谢和毒性特征,包括某些免疫抑制剂和抗癌药物。
6.UGT1A1(尿苷二磷酸葡萄糖醛酸转移酶 1A1)
尿苷二磷酸葡萄糖醛酸转移酶 1A1(UGT1A1)是一种关键的葡萄糖醛酸化酶,对于多种内源性和外源性化合物(包括化疗药物)的解毒和排泄至关重要。
在肿瘤学中,UGT1A1 基因在伊立替康的代谢中起着关键作用。UGT1A1 基因的多态性可显著影响药物代谢,进而影响药物的疗效和毒性。除此之外,UGT1A1 基因的遗传变异(如 * 28/*28 纯合基因型、*6/*28 杂合基因型)可能导致酶活性降低,进而造成戈沙妥珠单抗结合障碍,增加其毒性事件发生风险。
恩西地平依赖 UGT1A1 代谢,UGT1A1 的活性差异会直接影响其临床安全性,核心风险是 “高胆红素血症”:UGT1A1 活性不足(*28等位基因)时,恩西地平在体内蓄积,不仅会直接增加药物毒性,还会与 “胆红素” 竞争 UGT1A1 的结合位点 —— 原本就因酶活性低而排泄不畅的胆红素,会进一步排出受阻,导致 “药物性高胆红素血症”。因此,恩西地平在治疗前通过基因检测明确UGT1A1 基因型。
6.1 UGT1A1 与伊立替康代谢
伊立替康是一种前药,需通过羧酸酯酶酶促转化为其活性形式 SN-38,SN-38 可抑制拓扑异构酶 I,导致癌细胞 DNA 损伤。然而,SN-38 对正常细胞也具有毒性,其蓄积可能导致严重的不良反应,如中性粒细胞减少(中性粒细胞数量减少,增加感染风险)、腹泻和黏膜炎。为降低毒性,SN-38 需通过 UGT1A1 进行葡萄糖醛酸化灭活,转化为无活性代谢产物(SN-38G)后排出体外。
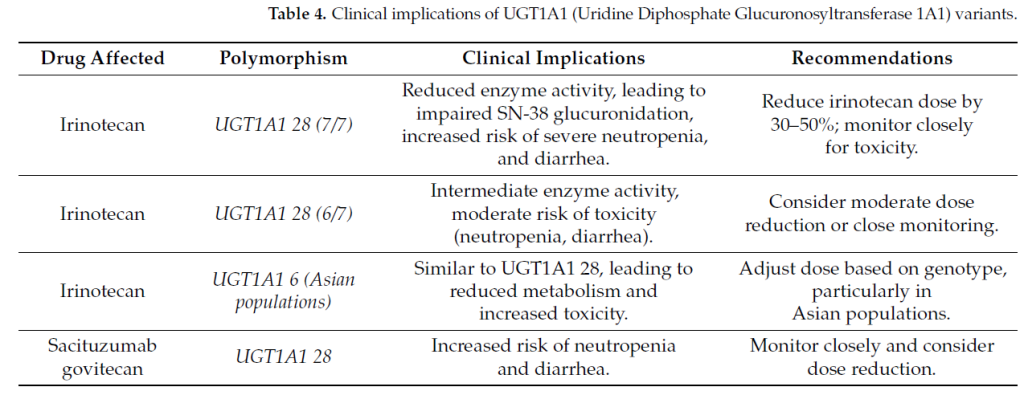
UGT1A128 多态性是 UGT1A1 基因中临床意义最显著的变异。该多态性涉及 TATA 盒启动子区域额外的 TA 重复序列(7 个重复序列,而通常为 6 个),导致转录活性降低和酶表达水平下降。
UGT1A128 纯合子患者(7/7 基因型)的 UGT1A1 活性显著降低,导致 SN-38 清除减少和药物暴露量增加。
杂合子携带者(6/7 基因型)的 UGT1A1 活性中等,导致 SN-38 葡萄糖醛酸化适度降低,其毒性反应风险虽低于纯合子携带者,但仍高于正常人群。
纯合子野生型基因型(6/6)患者的 UGT1A1 活性正常,在接受标准剂量伊立替康治疗时,发生伊立替康相关毒性反应的可能性较低。
6.2 UGT1A1 多态性在伊立替康治疗中的临床意义
伊立替康治疗最严重且剂量限制性的毒性反应之一是中性粒细胞减少。
UGT1A1*28 多态性的患者(尤其是 7/7 基因型患者),通常起始伊立替康剂量会降低 30%-50%,同时在治疗期间密切监测血常规和胃肠道症状,以预防严重毒性反应。
杂合子携带者(6/7 基因型)可能需要根据其临床耐受性和整体健康状况进行不太剧烈的剂量调整。
多个专业指南(包括临床药物基因组学实施联盟(CPIC)和欧洲药品管理局(EMA)的指南)建议在治疗前进行 UGT1A1 基因分型,尤其是对于接受高剂量(≥250 mg/m²)伊立替康治疗的患者。
除基因分型外,在特定临床场景中,可通过监测 SN-38 水平进行治疗药物监测(TDM),根据药物暴露量实时进行个性化剂量调整。
对于携带 UGT1A1 变异的患者,指南建议伊立替康起始剂量降低 30%,且禁止使用 240 mg/m² 或更高剂量的伊立替康。
6.3 UGT1A1 在其他癌症治疗中的作用
虽然伊立替康是受 UGT1A1 多态性影响最广泛认可的化疗药物,但其他肿瘤治疗药物也依赖 UGT1A1 介导的代谢,尽管程度较低。这些药物包括某些激素治疗药物(如雌激素和雄激素)以及一些新型靶向治疗药物。
3 期 ASCENT 试验评估了戈沙妥珠单抗(SG)在至少接受过两种化疗方案后复发或难治的转移性三阴性乳腺癌(mTNBC)患者中的安全性。在探索性安全性分析中,与其他基因型患者相比,UGT1A1*28/28 基因型患者发生 3 级及以上中性粒细胞减少、发热性中性粒细胞减少、贫血和腹泻的发生率更高。因此,接受戈沙妥珠单抗治疗的 UGT1A128/*28 基因型患者应受到密切监测。
6.4 研究与未来方向
多态性研究
目前正在进行的研究继续调查不同人群中的 UGT1A1 多态性。例如,UGT1A16 变异在亚洲人群中更为常见,与 UGT1A128 类似,可显著降低酶活性。了解这些人群特异性变异的频率和影响,对于制定针对不同种族群体的药物基因组学指南至关重要。
联合治疗影响
UGT1A1 多态性如何影响联合治疗的疗效和安全性,尤其是包含伊立替康与其他化疗药物或靶向药物的治疗方案。
其他同工酶
尽管 UGT1A1 是药物代谢相关研究最广泛的 UGT 家族同工酶,但其他同工酶(如 UGT1A4 和 UGT1A9)也可能参与药物代谢,目前正在研究它们对化疗结局和毒性的潜在影响。
7.GSTs(谷胱甘肽 S – 转移酶)
谷胱甘肽 S – 转移酶(GSTs)是一个酶家族,负责外源性物质(包括铂类化合物)的解毒。GSTs 分为多个类别,每类由不同的基因或基因家族编码。肿瘤患者接受环磷酰胺、卡铂、多柔比星、顺铂、奥沙利铂等药物治疗时,这些基因的多态性可能导致药物作用改变或毒性表现不同。
GST 由 GSTP1 基因编码,该基因中最常见的多态性是外显子 5 中的非同义单核苷酸多态性(313 A>G),与奥沙利铂治疗的更佳疗效相关。高加索人群中该多态性的检出率约为 40%-45%,亚洲人群中为 27%。
一项针对 107 例接受 5-FU / 奥沙利铂治疗的晚期结直肠癌患者的研究显示,携带变异基因型的患者与携带野生型基因型的患者相比,中位生存期存在显著差异:前者为 24.9 个月,后者为 7.9 个。
另一项针对 64 例接受奥沙利铂为基础治疗的胃肠道癌患者的研究发现,GST 活性降低与毒性增加相关,野生型基因型患者更易出现严重(3 级)神经病变。
一项关于非小细胞肺癌(NSCLC)患者谷胱甘肽 S – 转移酶基因多态性的荟萃分析显示,GSTM1 缺失型和 GSTP1 Ile105Val 基因 GG 基因型均与顺铂为基础化疗的更佳临床结局和治疗反应相关。GSTP1 Ile105Val 基因多态性在东亚 NSCLC 患者中比在高加索患者中更为常见。携带 GSTM1 缺失型多态性的中国 NSCLC 患者治疗反应更佳。然而,为进一步研究 GST 多态性的作用,未来需要开展更全面、种族多样化的人群临床试验。
DNA修复基因
DNA 修复基因通过修复药物诱导的 DNA 损伤影响肿瘤细胞敏感性,其多态性与化疗疗效密切相关。
1.MGMT(O-6 – 甲基鸟嘌呤 – DNA 甲基转移酶基因)
MGMT 是修复 DNA 损伤的关键酶。其修复功能对正常细胞过程至关重要,通过直接逆转烷基化损伤,MGMT 可使癌细胞对烷化剂类化疗药物产生耐药性。
替莫唑胺、达卡巴嗪等烷化剂类药物常用于治疗脑肿瘤、肉瘤及淋巴瘤,而癌细胞中 MGMT 的存在可能降低这些药物的疗效。
Stupp 试验证实了 MGMT 甲基化的预测性生物标志物作用:与 MGMT 未甲基化组相比,MGMT 甲基化的胶质母细胞瘤患者在标准放疗基础上加用替莫唑胺,生存期获益显著更高。
长期随访结果进一步证实,MGMT 基因启动子甲基化在预测替莫唑胺敏感性方面持续具有重要意义。
在胶质母细胞瘤中,MGMT 启动子甲基化既是预后标志物,也是预测患者对烷化剂类药物治疗反应的标志物。
2.ERCC1(切除修复交叉互补组 1)
ERCC1 基因参与核苷酸切除修复通路,且与含铂抗癌药物介导的基因特异性修复相关。
研究发现,膀胱癌患者中 ERCC1 水平升高与结局较差相关。而在非小细胞肺癌中,外显子 4 的多态性(496 C>T)与 ERCC1 活性降低相关,进而与更长的生存期相关。另一项针对 91 例接受 5-FU / 奥沙利铂治疗的晚期结直肠癌患者的临床试验探讨了上述多态性,结果显示,携带变异型纯合子的患者反应率显著高于杂合子患者(42.3%)和野生型基因型患者(21.4%),达到 61.9%。
总之,DNA 修复增强会降低铂类药物的疗效,但目前也存在一些矛盾结果,因此需要开展更多研究以证实该数据。
3.ERCC2(切除修复交叉互补组 2)
ERCC2 是另一个属于核苷酸切除修复通路的基因,已发现该基因存在多种单核苷酸多态性,其中最常见的包括 965 G>A(Asp321Asp)和 225 A>C(Lys751Gln),这些多态性与 DNA 修复能力降低相关。
一项针对 73 例接受 5-FU / 奥沙利铂治疗的晚期结直肠癌患者的研究显示,携带 225 A>C 多态性的患者临床结局和长期生存率较差:野生型基因型患者的平均生存期约为 17.4 个月,杂合子患者为 12.8 个月,纯合子患者仅为 3.3 个。
ERCC1 和 ERCC2 基因均对 DNA 修复系统至关重要,因此均参与核苷酸切除修复通路。2012 年针对中国人群开展的一项临床研究(纳入 213 例结直肠癌患者和 240 例无癌对照者)表明,这些基因的单核苷酸多态性与结直肠癌风险增加相关。研究对四种功能性单核苷酸多态性进行了基因分型:ERCC1 Asn118Asn、C8092A,ERCC2 Asp312Asn 和 Lys751Gln。结果发现,与 CC 基因型相比,携带 ERCC1 C8092A 多态性 AA 和 CA/AA 变异基因型的个体结直肠癌风险显著升高,而其他单核苷酸多态性未观察到与结直肠癌存在重要关联。因此,ERCC1 C8092A 多态性可能成为中国人群结直肠癌易感性的重要标志物,但在常规临床应用之前,需要开展更大规模的研究以证实这些发现。
4.XRCC1(X 射线修复交叉互补组 1)
DNA 修复蛋白 XRCC1 由人类 XRCC1 基因编码,参与电离辐射和烷化剂诱导的 DNA 单链断裂修复、碱基切除修复和核苷酸切除修复。这些机制会影响铂类抗癌药物的疗效。其中一种单核苷酸多态性(1301 G>A;Arg399Gln)会导致碱基切除修复能力突变、癌症发病风险增加以及晚期结直肠癌患者的治疗反应变差。
一项针对 61 例接受 5-FU / 奥沙利铂治疗的晚期结直肠癌患者的临床试验显示,73% 治疗反应良好的患者携带野生型基因型,而反应良好的患者中无一人为纯合子变异基因型。此外,研究发现携带该变异等位基因的非小细胞肺癌患者生存率显著降低。
分子研究表明,XRCC1 的单核苷酸多态性与多种癌症(如胃癌)的发病风险相关,且对治疗结局具有预测价值。一项纳入 612 例胃癌患者的研究采用免疫组织化学(IHC)方法评估了手术标本中 XRCC1 蛋白的表达谱,结果发现,XRCC1 免疫组织化学检测阴性的患者从铂类为基础的辅助化疗中获益更多。这些结果支持 XRCC1 阴性表达使肿瘤对铂类化疗药物更敏感的观点。检测胃癌患者的 XRCC1 表达可为选择最佳辅助治疗提供临床指导,但需要开展更多大规模研究以明确具体机制。
药物转运体基因
ABCB1、ABCC2、ABCG2(ATP 结合盒转运体)
ATP 结合盒转运体(又称 ABC 转运体)是一个超家族转运系统,利用 ATP 结合和水解产生的能量将多种底物跨细胞膜转运。其活性的任何改变都可能导致多种药物(包括化疗药物)的清除率发生变异。
ABCB1 基因(又称 MDR1,多药耐药基因)编码 P – 糖蛋白,该糖蛋白在对特定抗癌方案耐药的细胞中过度表达。
MDR1*2 单倍型中存在两个同义单核苷酸多态性(外显子 12 的 C1236T 和外显子 26 的 C3435T)和一个非同义单核苷酸多态性(外显子 2 的 G2677T)。该单倍型会导致 P – 糖蛋白上调、药物转运体活性增强以及 SN-38 清除率降低。
参与伊立替康代谢的其他药物转运体还包括 ABCG2 和 ABCC2。ABCG2 又称乳腺癌耐药蛋白,其特定的 421 C>A 变异与伊立替康的处置相关;而 ABCC2 的 ABCC22 单倍型与伊立替康治疗的不良反应(最常见为腹泻)减少相关。一项针对 167 例接受伊立替康治疗的患者的研究显示,携带 ABCC22 单倍型的患者中仅 10% 出现腹泻,而其他患者中这一比例为 44%。
现有数据表明,包括 ATP 超家族在内的特定药物转运体是药物耐药性和反应改变的关键因素。
二
药物基因组学在肿瘤临床实践中的特殊考量
癌症化疗面临的最主要问题是耐药性的产生和严重的不良反应。由于大多数抗癌药物不具有肿瘤特异性,因此也会对正常细胞造成损伤,这使得无法使用可能根除敏感性较低的肿瘤细胞群体所需的高剂量药物。治疗反应的个体差异可以通过每个人特有的遗传变异来解释。
药物遗传学的进展可能成为改变癌症治疗的关键。将患者基因分型引入临床环境有助于制定化疗方案和药物剂量决策,以实现最大疗效和最小毒性风险。
如果未来能够从多项临床试验中收集到充分且可靠的数据,药物基因组学最实际的应用之一将是在存在多种等效治疗方案的疾病情况下,帮助选择最合适的方案。未来,临床医生很可能需要谨慎评估特定药物的风险 – 获益比和药物基因组学概率,以确保为减少不良反应或预防毒性而进行的剂量调整或药物替换不会影响治疗结局。
参考资料
Erika Cecchin, Gabriele Stocco. Pharmacogenomics and Personalized Medicine. Genes (Basel). 2020 Jun 22;11(6):679
Wolfgang Sadee, et al. Pharmacogenomics: Driving Personalized Medicine. Pharmacol Rev. 2023 Jul;75(4):789-814
Ramón Cacabelos, et al. Genophenotypic Factors and Pharmacogenomics in Adverse Drug Reactions. Int. J. Mol. Sci. 2021, 22(24), 13302.
Rodrigo Sánchez-Bayona, et al. Pharmacogenomics in Solid Tumors: A Comprehensive Review of Genetic Variability and Its Clinical Implications. Cancers 2025, 17(6), 913.
Yucai Jiang, Guolin Alexander Wen. Deciphering gene mutations in the efficacy and toxicity of antineoplastic drugs: an oncology pharmacist’s perspective. Front. Pharmacol. 20 March 2025. Volume 16.
Nelly N. Miteva-Marcheva, et al. Application of pharmacogenetics in oncology. Biomarker Research (2020) 8:32