


人全外显子液相基因芯片(WES)临床应用的进展-心血管疾病(上)
- boke
- 2025-06-19
- 4:32 下午
心肌病
心肌病是一组异质的心肌疾病,可定义为存在心肌结构和(或)功能异常,且用高血压、冠状动脉粥样硬化性心脏病、瓣膜性心脏病和先天性心脏病不足以解释其病因的心肌疾病。
心肌病可分为心室心肌病和心房心肌病,前者包括肥厚型心肌病(hypertrophic cardiomyopathy,HCM)、 扩张型心肌病(dilated cardiomyopathy,DCM)、限制型心肌病(restrictive cardiomyopathy,RCM)、心律失常相关心肌病、代谢性心肌病、综合征性心肌病、淀粉样变心肌病、其他心肌受累疾病和特异性心肌损伤 9 大类。
最常见的是扩张型心肌病(DCM)和肥厚型心肌病(HCM)。我国针对普通人群的抽样调查数据显示,成人 HCM 患病率约为1/625。近年来,随着影像和遗传诊断技术发展,HCM 的患病率估计可达1/200。DCM 患病率目前尚缺乏基于普通人群的流行病学调查结果,估计 DCM 的患病率与 HCM 相当。西方人群中,致心律失常性右心室心肌病(ARVC)的患病率约为1/5000~1/2000。其他心肌病尚缺乏流行病学数据。考虑到认知和诊断方法的限制,心肌病的患病率可能被严重低估。
1. 大规模全外显子测序,发现辅助蛋白 BAG3 C151R 变异对心肌病的双向调节作用-保护 DCM 以及TTN 介导的 DCM,但增加 HCM 风险。
文献标题:Bidirectional Risk Modulator and Modifier Variant of Dilated and Hypertrophic Cardiomyopathy in BAG3
发表期刊:JAMA Cardiology
发表时间:2024年

背景:罕见的基因变异与 DCM 和 HCM 的发生有关,这些基因编码心脏肌节蛋白、核蛋白、离子通道蛋白、细胞骨架蛋白等。遗传性 DCM 具有年龄依赖性、外显率降低和可变表达性,表现出多种表型,从早期症状(如心律失常或传导障碍、轻度心室功能异常)到明显的扩张型心肌病。这在与罕见致病性 TTN 截短变异相关的 DCM 中得到证实,这些变异是遗传性 DCM 最常见的基因病因之一。然而,影响 TTN 截短变异在 DCM 中可变外显率的基因修饰因子在很大程度上仍不清楚。
BAG3 的罕见功能丧失变异是 DCM 的罕见原因,估计占 DCM 病例的0.3%。BAG3 基因常见变异与 DCM 和 HCM 均有关联。与大多数其他心肌病相关基因不同,BAG3不编码肌节、核蛋白、离子通道蛋白或细胞骨架蛋白。相反,BAG3 编码一种广泛表达的细胞蛋白,在心肌细胞中发挥多种作用,包括与热休克蛋白家族成员协同作用,清除错误折叠和降解的蛋白;通过与 BCL2 相互作用抑制细胞凋亡;并维持肌节Z带的结构完整性。鉴于 BAG3 与 DCM 和 HCM 的关联,以及其在应激状态下稳定心肌细胞的多重功能,BAG3 可能包含 DCM(包括 TTN 介导的 DCM )的潜在修饰变异。
方法:这是一项在宾夕法尼亚大学医学生物样本库 (PMBB) 进行的横断面研究,纳入了 2014 年至 2023 年宾夕法尼亚大学医学院临床实践站点患者。全外显子组测序 (WES) 与电子病历 (EHR) 数据结合,分析 BAG3 编码区变异与 EHR 表型之间的关联。这是一项基于医疗保健人群的研究,包括 PMBB 中具有 WES 与 EHR 表型关联的欧洲和非洲遗传血统个体,并在 BioVU、UK Biobank、MyCode 和 DCM 精准医学研究中进行了重复研究。重要的是,我们评估了 BAG3 常见错义变异与 DCM 和 HCM 的关联,以及其对 TTN 介导的 DCM 结局的影响。
结果: 在PMBB数据库 (n = 43 731)的 30 324 名欧洲人和 11 198 名非洲人中,常见的 C151R 变异与 DCM 风险降低 (OR=0.85) 和同时 HCM 风险增加 (OR=1.59) 相关,这在重复研究中得到了证实。与非携带者相比,C151R 携带者在纵向结果方面有所改善,例如死亡年龄 (HR=0.85;携带者中位年龄为 71.8 岁,非携带者中位年龄为 70.3 岁) 和心脏移植 (HR=0.81;携带者中位年龄为 56.7 岁,非携带者中位年龄为 55.6 岁)。在具有高心脏表达的 TTN 外显子截短变异 (n=358) 的个体中,C151R 与 DCM 风险降低 (OR=0.42) 和心力衰竭风险降低 (OR=0.27) 相关。
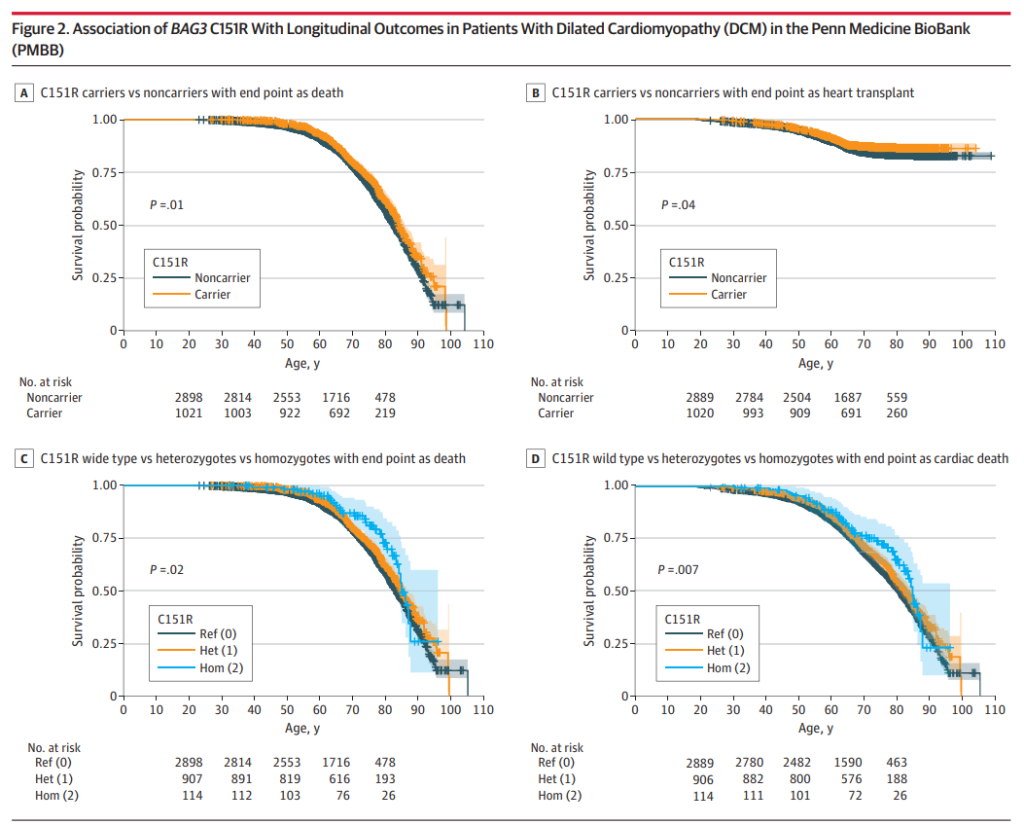
结论与意义:BAG3 C151R 被确定为 DCM-HCM 谱系风险的双向调节因子,以及 TTN 介导的 DCM 中的重要遗传修饰变异。这项研究扩展了对 DCM 病因和外显率的理解,表明 BAG3 C151R 是导致 DCM 可变表达性的重要遗传修饰变异,值得进一步探索其机制以及更广泛的 DCM 遗传修饰因素。
2. 通过对一个 DCM 家系的 WES 测序,发现新型致病变异并阐明其分子机制,同时为 DCM 提供了潜在治疗策略。
文献标题:Disruption of cTnT-Mediated Sarcomere–Mitochondrial Communication Results in Dilated Cardiomyopathy
发表期刊:Circulation
发表时间:2025年

背景:扩张型心肌病(DCM)在很大程度上受到遗传因素的影响。肌节功能与其他细胞器密切相关,特别是肌节与线粒体之间的相互调节。线粒体应激失调与 DCM 的进展有关,但其机制尚不完全清楚。
方法:通过对一个DCM家系的人全外显子测序(WES),在 cTnT(TNNT2)中发现了一种新的致病变异(p.K185E)作为致病变异。通过分析 iPSC 衍生的心肌细胞、心脏类器官以及小鼠的病理表型,来阐明其分子机制。同时,使用RNA测序、代谢物谱分析和免疫共沉淀质谱分析技术来揭示这些机制。
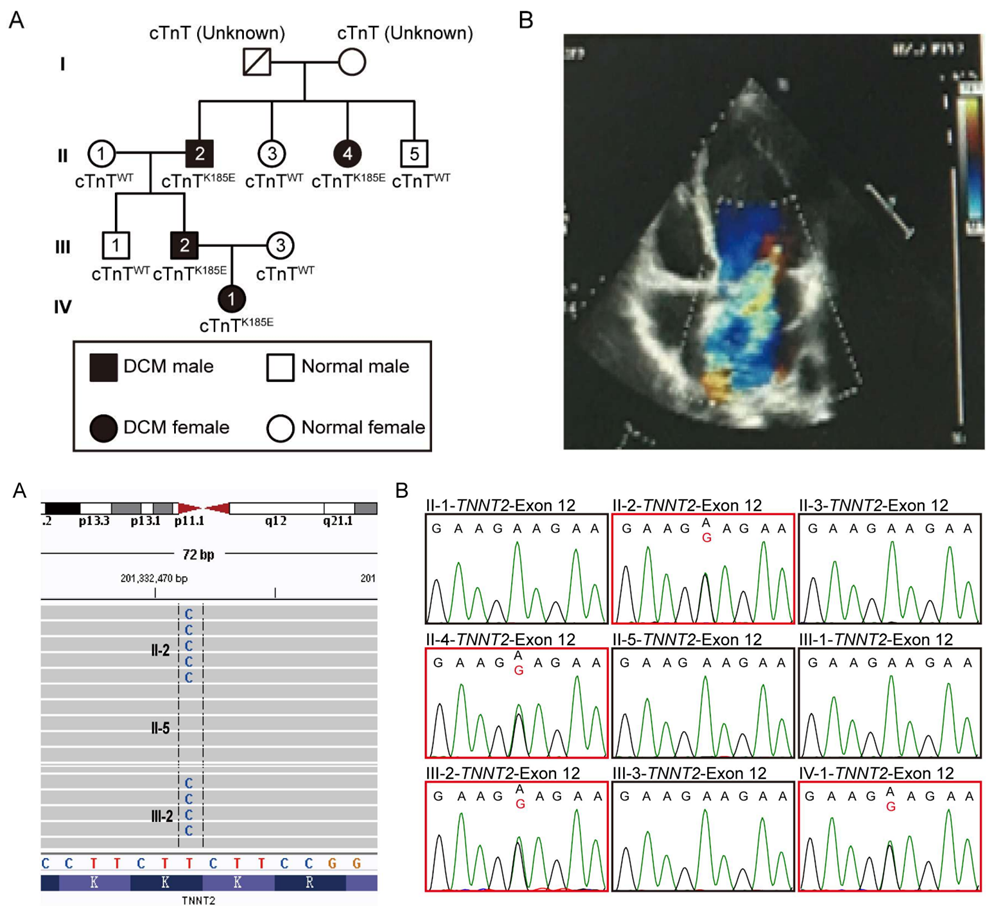
结果:在来自 DCM 患者的 iPSC 衍生心肌细胞中,观察到肌小节紊乱和线粒体碎片化,并伴有严重的线粒体功能障碍。cTnT(p.K185E)和14-3-3蛋白之间的相互作用减弱,导致14-3-3蛋白从肌小节结构中分离。游离的14-3-3蛋白异常地参与RAS/RAF1信号轴,驱动异常的p44/42激酶激活,最终导致线粒体裂变调节剂DRP1(动力学相关蛋白1)和MFF(线粒体分裂因子)的磷酸化。这些观察结果在iPSC衍生的心脏类器官中得到了重复。
携带同源cTnT序列变异的敲入小鼠也表现出人类DCM的标志性特征,包括心脏功能障碍、心室扩张、肌小节紊乱和线粒体碎片化。此外,Mdivi-1,一种线粒体分裂抑制剂,在体内缓解了DCM表型。
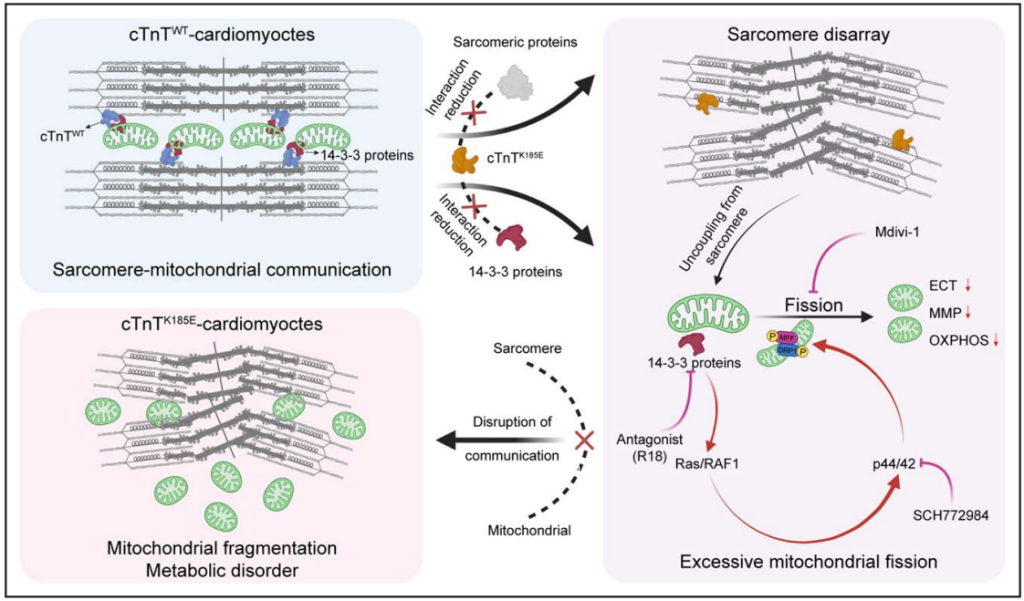
结论:我们的研究结果描绘了 DCM 背后的一种新的致病机制,证明了cTnT(p.K185E)序列变异通过削弱cTnT和14-3-3蛋白之间的相互作用来破坏肌小节-线粒体通信,从而通过过度激活14-3-3蛋白介导的RAS/RAF1-p44/42-DRP1/MFF信号轴来加速线粒体碎片化。因此,针对14-3-3蛋白和p44/42激酶活性的治疗可能是治疗 DCM 和其他与线粒体动力学异常相关的心脏疾病的一个有前途的策略。
先天性心脏病
先天性心脏病(简称先心病)是指胚胎发育异 常导致的心脏和胸腔内大血管的结构和功能异常。 先心病是我国最常见的出生缺陷,发病率为 8.98%。先心病病因复杂,多数是环境因素和遗传因素共同作用所致。研究报道约1/3的先心病存在遗传因素,其中染色体数目异常约占10%,染色体结构畸变占5%~20%,单基因变异约占10%。
1. 321 名 CHD 先证者单样本全外显子测序重分析,揭示NODAL变异的潜在致病机制
文献标题:NODAL variants are associated with a continuum of laterality defects from simple D-transposition of the great arteries to heterotaxy
发表期刊:Genome Medicine
发表时间:2024年

背景:单基因缺陷目前可以解释约 10% 的先天性心脏病,大多数致病/可能致病变异为新发变异。很多 CHD 都跟左右不对称有关,左右不对称病因的遗传基础很复杂,我们对相关基因的了解还很有限,但常染色体显性、常染色体隐性和X连锁遗传模式在涉及异位的罕见病性状中均有观察到。
NODAL信号通路是左右轴发育的分子控制中的关键参与者,在脊椎动物胚胎和心脏发育中起着至关重要的作用。基因突变导致TGF-β/NODAL信号通路紊乱,已被证实会导致人类的内脏异位症。
为了进一步探索这种关联并改善分子诊断,研究人员在一个大样本的左右不对称个体中,找到了更多不同的NODAL变异,然后详尽的研究了它们的潜在致病机制。
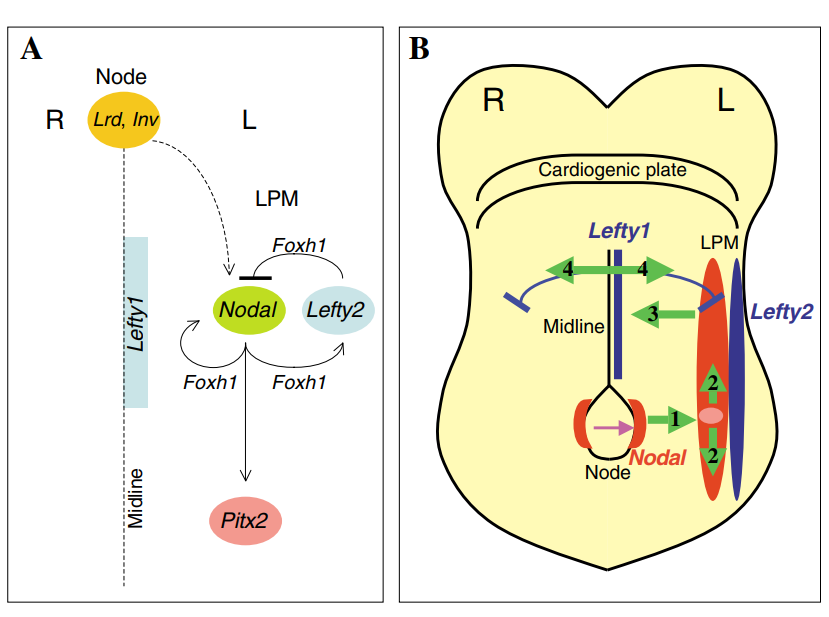
方法:该研究使用基于家系的罕见变异基因组分析,重新分析了321名患有临床诊断的偏侧性先天性心脏病 (CHD) 的先证者单样本全外显子测序数据,并向该队列添加了 12 名携带已知 NODAL 变异的 CHD 受试者,以调查等位基因类型。
Sanger 测序用于等位基因确认和遗传分离分析。Array-CGH 和 ddPCR 用于拷贝数变异 (CNV) 验证和表征。基于人类表型数据库的定量表型分析,以剖析等位基因特异性表型差异。
结果:在 33 例 CHD 病例中,NODAL 的错义、无义、剪接位点、插入缺失和/或结构变异被确定为异位和其他外侧性缺陷的潜在原因。已鉴定的两个 NODAL 错义变异(c.2T>C,p.M1T;c.1039C>T,p.L347F)代表未报道的变异等位基因,以前从未与偏侧性缺陷相关联。
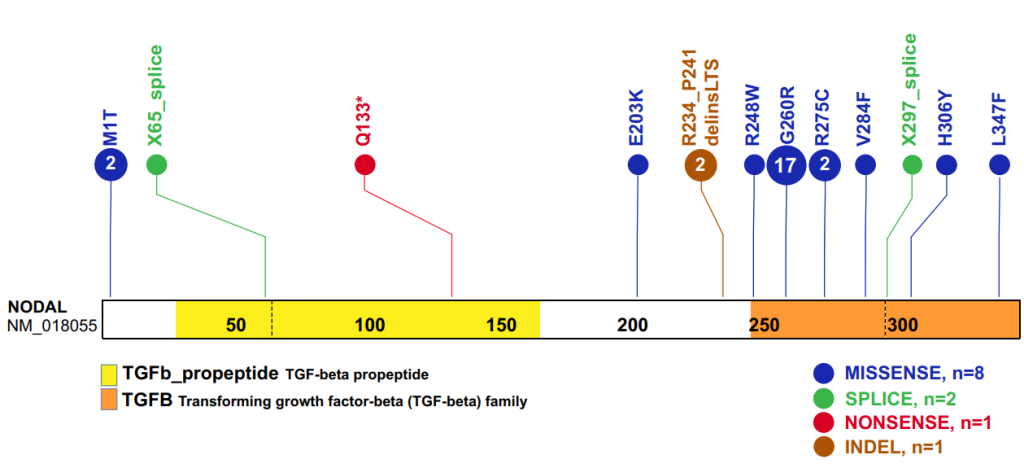
该研究在两个无关的 CHD 病例中发现了两个跨越 NODAL 的 CNV 缺失等位基因。此外,发现 17 名 CHD 个体(其中 16/17 名具有已知的西班牙裔血统)具有 c.778G>A:p.G260R NODAL 错义变异,因此建议将其从不确定意义的变异 (VUS) 重新分类为可能致病。研究还发现了一种反复出现的复杂插入缺失变异,其DNA二级结构预测暗示二级结构诱变是一种可能的形成机制。
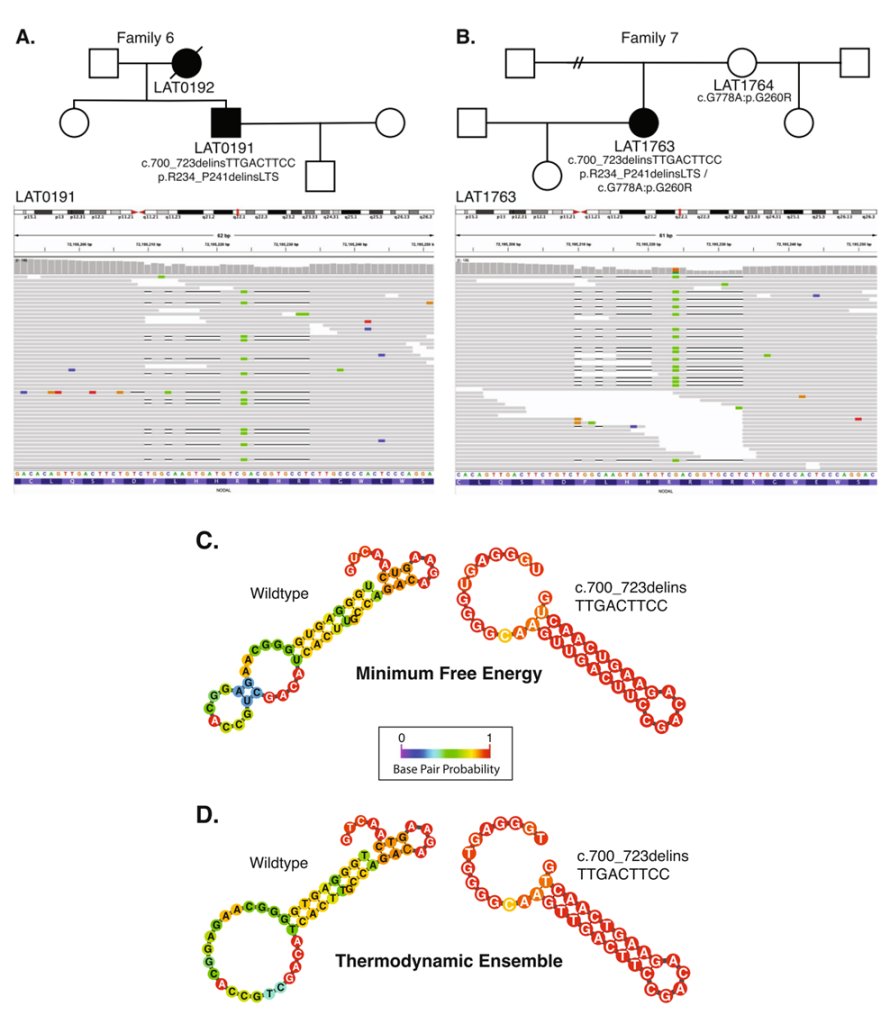
2. 大规模全外显子测序,解析CHD中隐性致病变异负担
文献标题:Recessive genetic contribution to congenital heart disease in 5,424 probands
发表期刊:PNAS
发表时间:2025年

背景:CHD 占所有出生缺陷的三分之一左右,全球新生儿发病率为1%至1.8%,所有年龄段患病率为0.16%。通过手术和导管介入治疗结构性心脏畸形的姑息治疗和修复,以及医疗管理的改善,使得近90%的 CHD 患者能够存活至成年,目前美国有240万人患有 CHD,其中包括140万成年人。尽管取得了显著进步,但包括心内膜炎、心律失常、再次手术、心力衰竭、肺动脉高压和神经发育缺陷在内的多种并发症或合并症使 CHD 成为一种常见的终身疾病。
CHD具有强烈的遗传基础。约25%的病例与大型染色体异常或拷贝数变异相,许多单基因变异也与 CHD 相关。隐性基因型(RGs)通常通过近亲家系和候选基因分析得到。尽管如此,对最常见的 CHD 形式遗传贡献的合理估计,这些形式通常为散发性,只有在过去十年中随着NGS测序技术的出现和 CHD 先证者及其父母样本的无偏收集才成为可能。
结果:我们分析了来自NHLBI Bench-to-Bassinet项目的5,424例 CHD 先证者的全外显子测序结果。罕见的破坏性的隐性基因型(RGs)估计至少导致2.2%的 CHD,相比于其他亚组(1.4%),其在偏侧表型中更多富集(5.4%)。
在108个经过筛选的人类隐性 CHD 基因中,发现了66个RGs,位于23个基因中,而85个基因没有RG。其中54 RGs个位于11个的基因中(有>1个RG),剩余12个基因有1个RG。近亲家系后代的RGs(4.7%,32/675)比在非近亲先证者(0.7%,34/4749)更为普遍。在410名阿什肯纳兹犹太先证者中,GDF1 和 PLD1 中的创始变异占RGs贡献的 74 %。
我们鉴定出 C1orf127 基因中存在广泛的RGs富集,该基因编码一种可能分泌的蛋白,在胚胎小鼠的脊索中表达,并与偏侧缺陷相关。
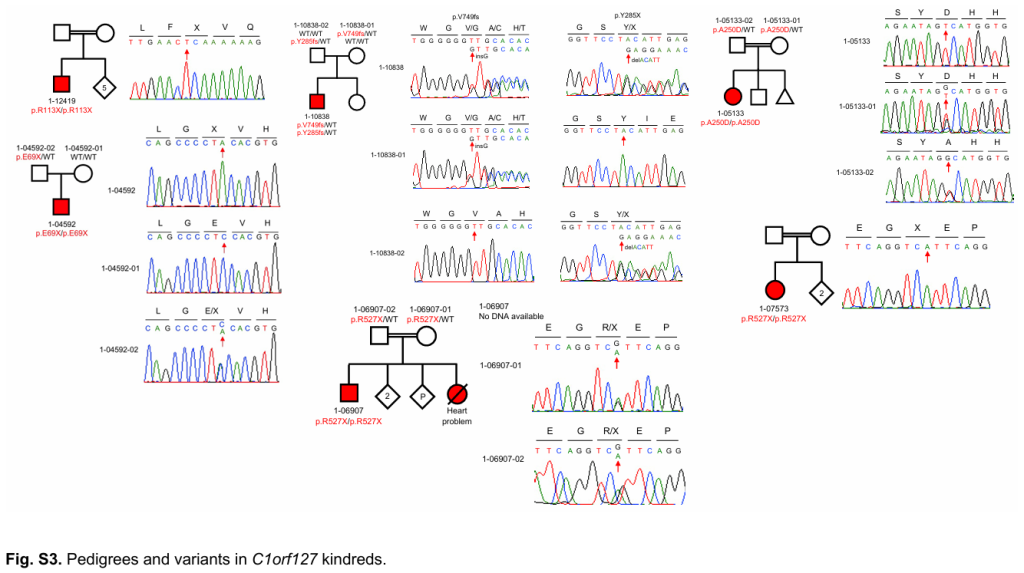
单细胞转录组揭示了在心肌细胞谱系中高度表达的基因中存在RGs的富集,包括在left-sided CHD患者中 MYH6、UNC45B、MYO18B 和 MYBPC3 等与收缩性相关的基因,支持异常收缩功能与这些缺陷的关联。具有显著RG负担的基因占患者总数的1.3%,超过推断总数的半数。这些结果展示了隐性基因型对 CHD 的贡献,因为每种基因可能只占总数的一小部分,表明许多基因仍有待发现。
理解先天性心脏病的遗传基础,有助于深入理解心脏发育过程,并对这一最常见的先天性疾病的预防、诊断和治疗提供启示。
TargetCap® Core Exome Panel v3.0
TargetCap@ Core Exome Panel v3.0基于伯科高品质DNA探针合成技术开发,全流程国产制造,由~40万条探针组成,以GRCh38/hg38人类参考基因组设计,参考Refseq、CCDS、ClinVar等数据库,覆盖19,524个基因,目标区域为33.9Mb。
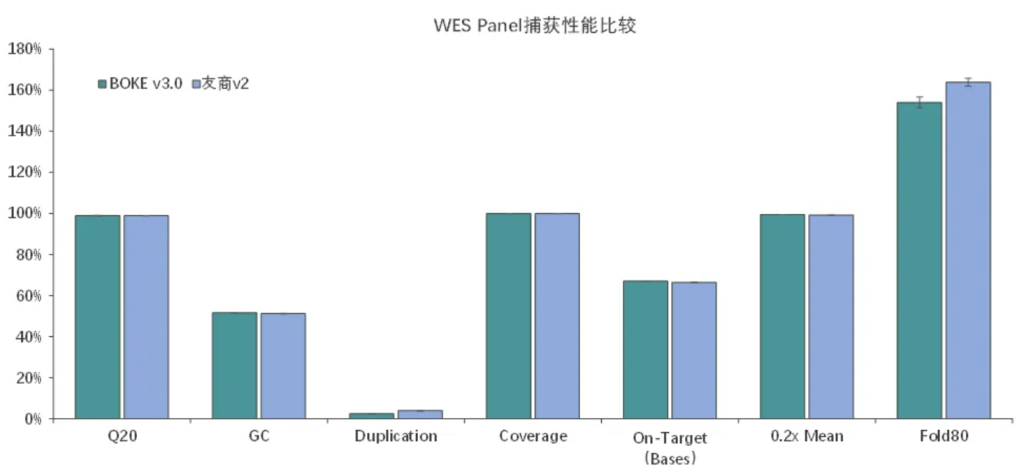
捕获性能比较
伯科全外芯片v3.0性能优异与国外友商同类型产品v2相当,中靶率、覆盖率、覆盖均一性等参数均达到国际领先水平。
适配高通量流程平台
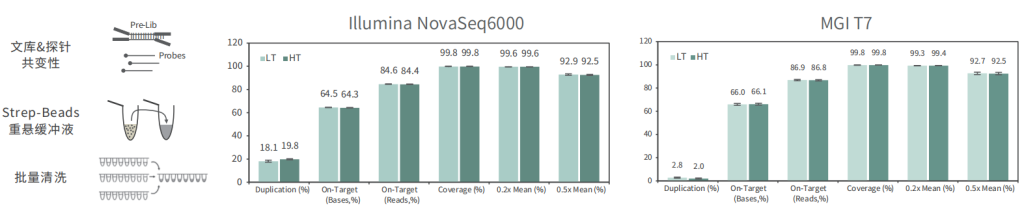
批次稳定
使用不同批次TargetCap® Core Exome Panel v3.0芯片对NA12878 gDNA进行捕获测序,结果显示,不同批次芯片在不同测序平台上均显示出优异的稳定性,不同位点的相对深度相关性高,批次稳定。
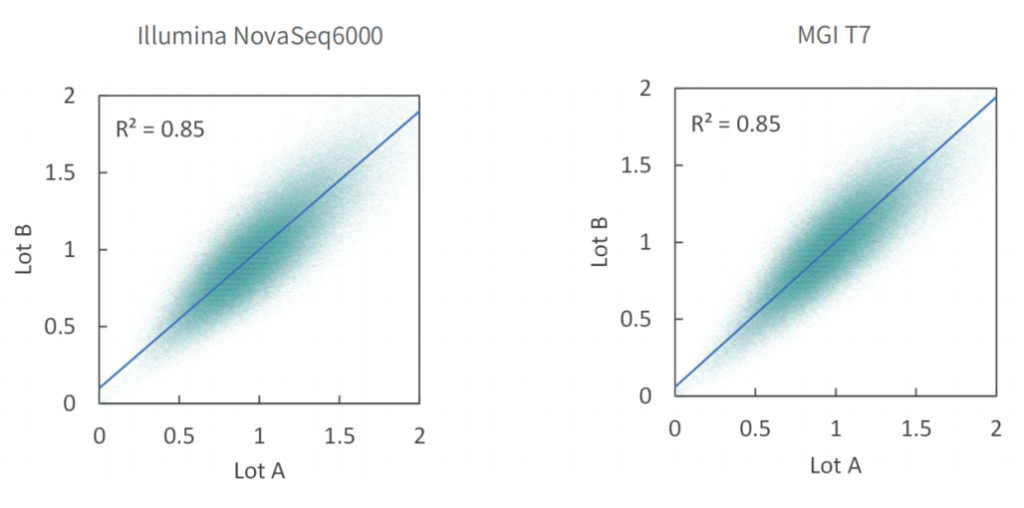
变异检测准确
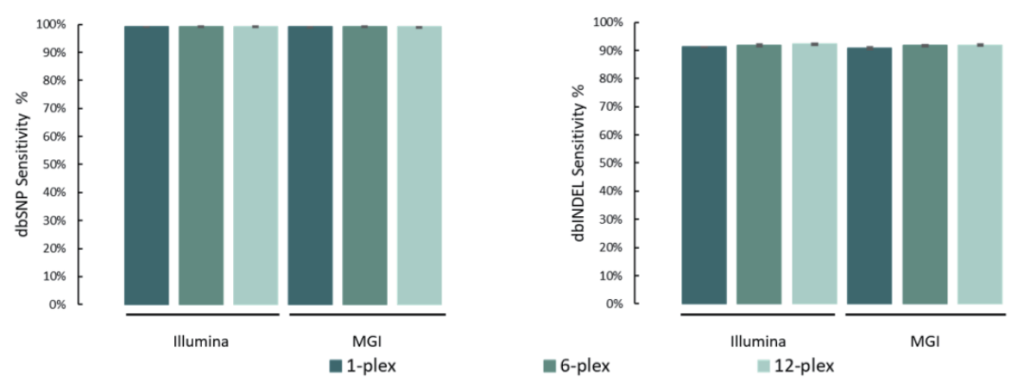
单核苷酸变异(SNV)和插入缺失 (INDEL)是基因组变异的常见形式,也是引起人类疾病的重要原因。
选取NA12878标准品,与预期SNV和INDEL变异进行比较。结果表明,在MGI与Illumina测序平台,SNP灵敏度为99.1%,INDEL灵敏度为91.6%。
添加线粒体模块临床样本表现
20例全血样本(S1#-S20#),采用1-4 Plex方式使用TargetCap® Core Exome Panel v3.0添加线粒体模块进行过夜杂交捕获;其中,S1#-S10#在Illumina NovaSeq X Plus平台测序, S11#-S20#在MGI MGISEQ-T7平台测序,均采用150PE模式测序。得到测序数据后,抽取8Gb数据进行生信分析。
两种测序平台的数据表现相近,平均深度分别为111x/115x (Illumina/MGI),中靶率优异均> 85%,覆盖均一性极佳(0.2X_MD≥99.3%);仅使用8Gb数据,高达98.5%的捕获区域达到了30X以上,99.5%的捕获区域达到20X以上,为临床样本检测提供了可靠的捕获数据。
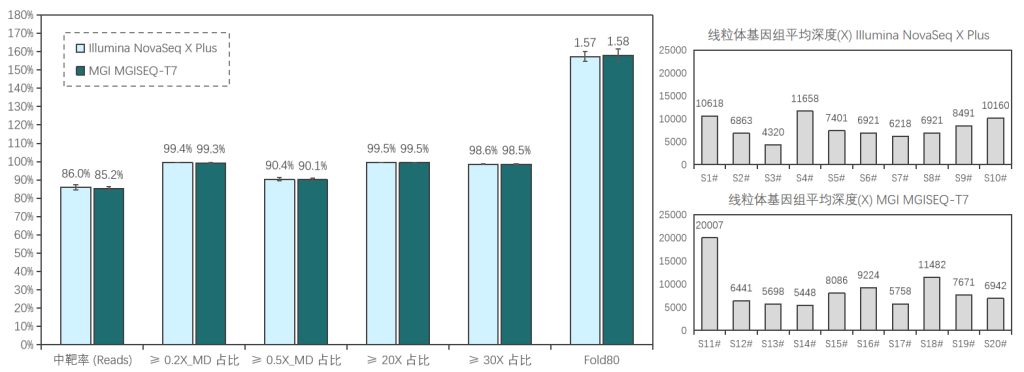
≥0.2X/0.5X_MD: Mean Depth,覆盖深度≥平均深度的0.2/0.5倍深度的区域占总区域的比例,用于表征覆盖均一性性能,越接近100%越好。
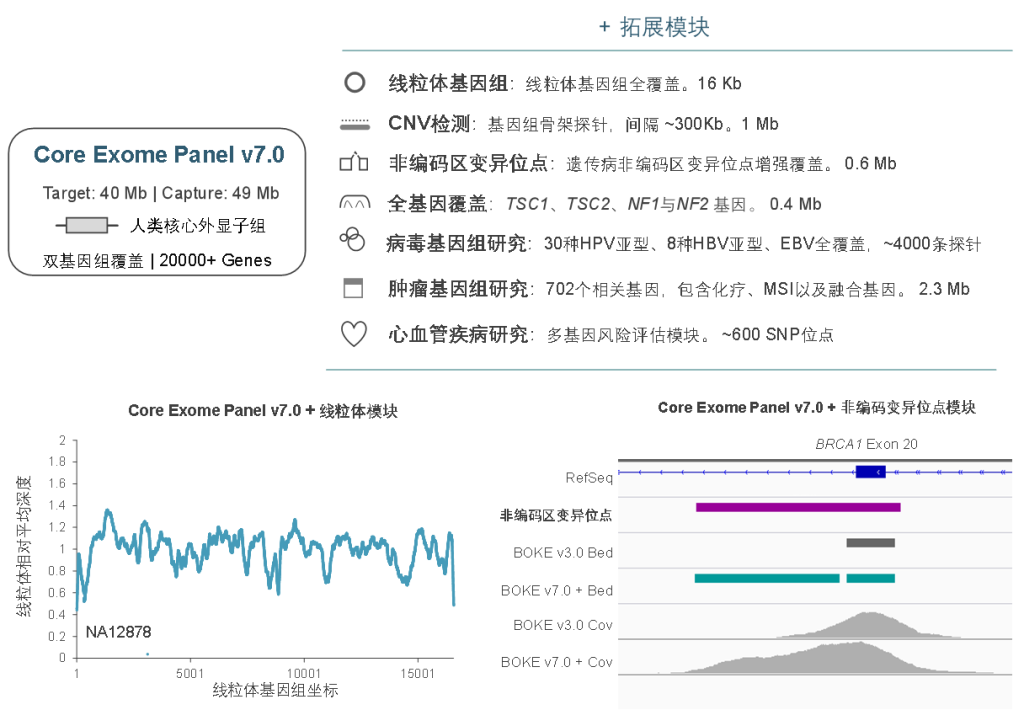
TargetCap® Core Exome Panel v7.0
TargetCap®Core Exome Panel v7.0(下文简称BOKE v7.0),该WES Panel增强了基因组hg19传统研究区域的覆盖,兼顾 hg19 & hg38 双版本基因组,可以更好的保证临床科研与转化的延续性。目标区域和捕获区域大小分别为40Mb和49Mb,对 hg19 传统研究区域覆盖提升至99.7%(友商I-v1),hg38传统研究区域覆盖相近(友商A-v8)。同时,新添加数百个具有一定功能与表型的基因,总基因数量达到20000+。
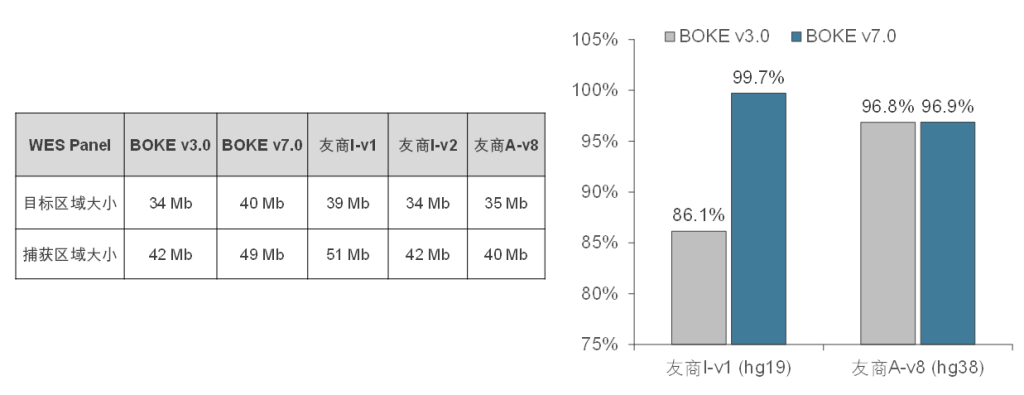
TargetCap® Core Exome Panel v7.0目标区域大小以及对不同友商产品目标区域的覆盖情况
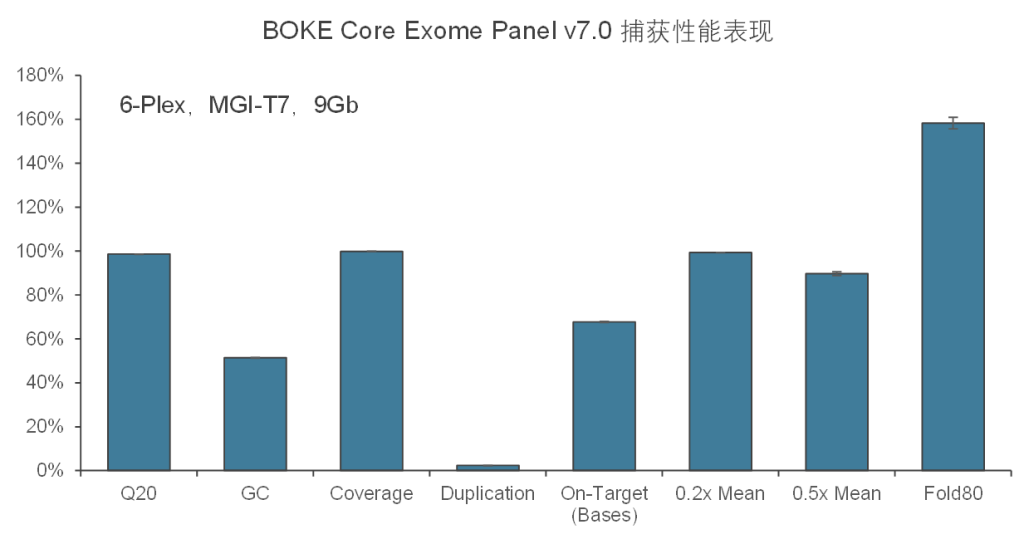
在捕获性能方面,TargetCap® Core Exome Panel v7.0依然表现优异,与TargetCap® Core Exome Panel v3.0表现相近。在测序9Gb条件下,平均深度达到110x左右,20x和30x以上区域占比分别为99.5%和98.5%,Fold 80为1.5-1.6之间,与国际领先产品数据表现相当。
同时,TargetCap® Core Exome Panel v7.0也可灵活的与拓展模块组合使用,满足不同场景的临床研究的需求及转化应用,包括线粒体、遗传病非编码区变异位点、单基因全覆盖、病毒基因组、肿瘤全景变异检测、重大疾病多基因风险评估模块等。此外,伯科公司自研自造的寡核苷酸合成平台可以快速响应个性化定制的需求,为人类基因组分子遗传学的研究与转化,提供更加全面高效的解决方案。
参考资料
1. 中国心肌病综合管理指南. 国家心血管病中心心肌病专科联盟; 中国医疗保健国际交流促进会心血管病精准医学分会. 中国循环杂志 2025 年 5 月 第 40 卷 第 5 期
2. Park J, Levin MG, Zhang D, et al. Bidirectional Risk Modulator and Modifier Variant of Dilated and Hypertrophic Cardiomyopathy in BAG3 [published correction appears in JAMA Cardiol. 2025 Feb 26. doi: 10.1001/jamacardio.2025.0076.]. JAMA Cardiol. 2024;9(12):1124-1133.
3. Ye L, Liu J, Lei W, et al. Disruption of cTnT-Mediated Sarcomere-Mitochondrial Communication Results in Dilated Cardiomyopathy. Circulation. Published online May 27, 2025.
4. 儿童先天性心脏病遗传学检测与临床应用专家共识(2025). 中华医学会儿科学分会心血管学组;中华儿科杂志编辑委员会. 中华儿科杂志2025年2月第63卷第2期
5. Dong W, Jin SC, Sierant MC, Lu Z, Li B, Lu Q, Morton SU, Zhang J, López-Giráldez F, Nelson-Williams C, Knight JR, Zhao H, Cao J, Mane S, Gruber PJ, Lek M, Goldmuntz E, Deanfield J, Giardini A, Mital S, Russell M, Gaynor JW, Cnota JF, Wagner M, Srivastava D, Bernstein D, Porter GA Jr, Newburger J, Roberts AE, Yandell M, Yost HJ, Tristani-Firouzi M, Kim R, Seidman J, Chung WK, Gelb BD, Seidman CE, Lifton RP, Brueckner M. Recessive genetic contribution to congenital heart disease in 5,424 probands. Proc Natl Acad Sci U S A. 2025 Mar 11;122(10):e2419992122.
6. Shiratori H, Hamada H. The left-right axis in the mouse: from origin to morphology. Development. 2006;133(11):2095-2104.
7. Dardas Z, Fatih JM, Jolly A, et al. NODAL variants are associated with a continuum of laterality defects from simple D-transposition of the great arteries to heterotaxy. Genome Med. 2024;16(1):53. Published 2024 Apr 3.