
个体化癌症疫苗的开发需要通过比较来自患者肿瘤和正常组织(比如白细胞)的NGS测序数据(基因组外显子测序和转录组测序)来识别肿瘤特异的蛋白质编码基因的非同义突变,但并不是每个突变都会引起免疫反应。
临床研究中研究最多的突变类型是编码区的单核苷酸变异(SNV)。未来的一个重要领域是新抗原发现工具的开发,这些新抗原是由癌症特异性INDELs、融合基因和剪接变异体产生的,这些新抗原与自身抗原的相似性低于SNV衍生的新抗原。
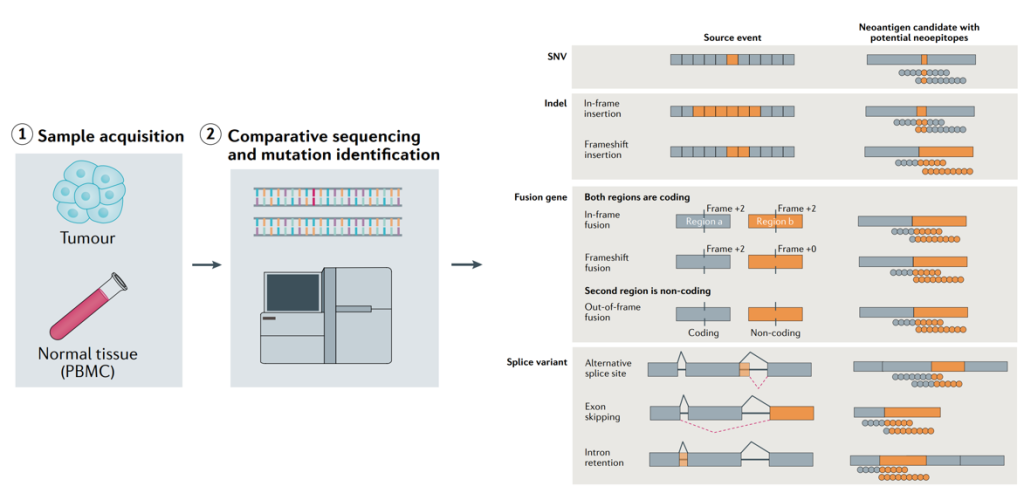
图1 肿瘤患者个体化突变信息获取
由于内含子区域通常较大,又常含有重复序列,因此,在基因组层面检测融合基因的性价比可能不高。基因融合和可变剪接事件检测在转录组测序中更容易处理,因此,转录组测序除了进一步确认基因组变异和基因表达量信息外,还可以用于基因融合和可变剪接事件检测。
对于转录组测序,mRNA-Seq(PolyA)和去核糖体RNA-Seq(Ribo-Zero)在处理临床样本中存在一定的局限性,mRNA-Seq流程较为简单,但其性能表现严重依赖样本质量,对于降解严重的RNA样本,其基因覆盖会受到明显影响;虽然Ribo-Zero对基因的覆盖均一性优于mRNA-Seq,但Ribo-Zero去核糖体效率与样本质量有关,同时还存在RNA前体干扰(Intron),其有效数据占比(Exon)往往较低,同时会因为样本质量出现波动,影响实验流程的稳定性(图2)。
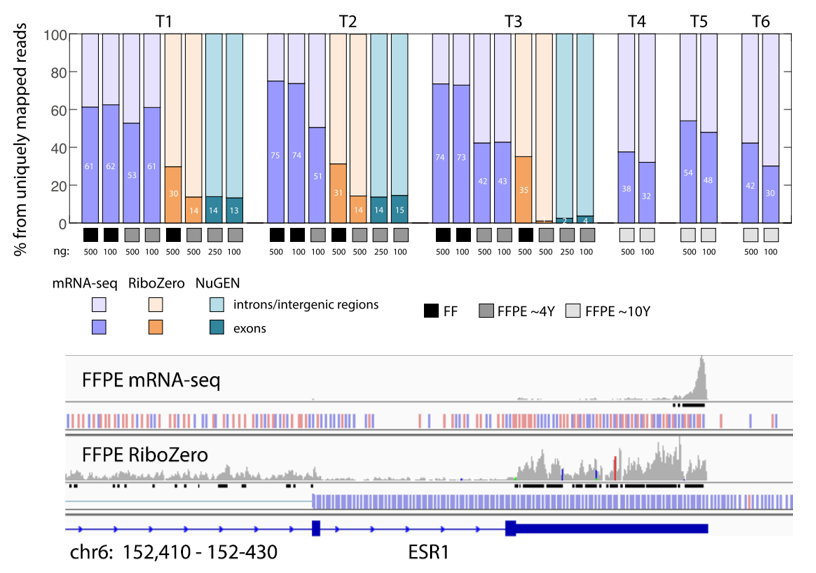
图2 mRNA-Seq和Ribo-Zero方法对不同类型、质量样本的测序表现[2]
转录组靶向捕获测序(RNA-Cap)不但耐受不同质量RNA样本,同时具有优异的基因覆盖均一性,是一种更加稳定、经济的转录组检测方法[3-6]。伯科全外显子芯片(Core Exome Panel v3.0)由120nt长链DNA探针组成,针对基因组序列设计,由于具有优异的GC和长度耐受能力,Core Exome Panel v3.0其对转录本序列同样具有良好的富集效果(图3)。
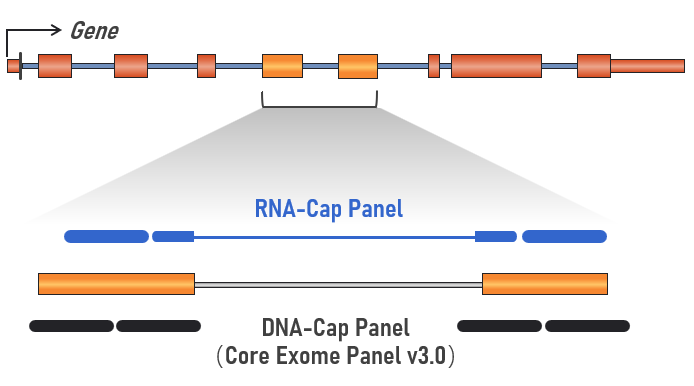
图3 Core Exome Panel v3.0探针设计示意图
利用肿瘤野生型和融合标准品样本(RIN:7.99/2.88),我们初步测试了伯科全外显子芯片(WES Cap)对于RNA样本的靶向捕获和融合基因检测表现。如图4所示,Total RNA-Seq和WES Cap分别测序108/115Gb和6.2Gb,按照WES Bed区间进行统计,Total RNA-Seq和WES Cap去Duplication后的测序深度分别为60.6x/72.1x和109-110x;覆盖率方面,由于针对基因组序列设计探针,WES Cap略低于Total RNA-Seq 2-4%;与Total RNA-Seq相比,WES Cap的On-Target(Reads)为93-96%,富集18-34倍,富集效率与先前RNA-Cap的报道一致[7],Gene Panel的目标基因越多,其富集效率越低。
此外,WES Cap的Exonic占比为72.9-76.3%,呈现经典靶向富集表现。在转录本覆盖均一性上,WES Cap与Total RNA-Seq相似,无明显偏好。
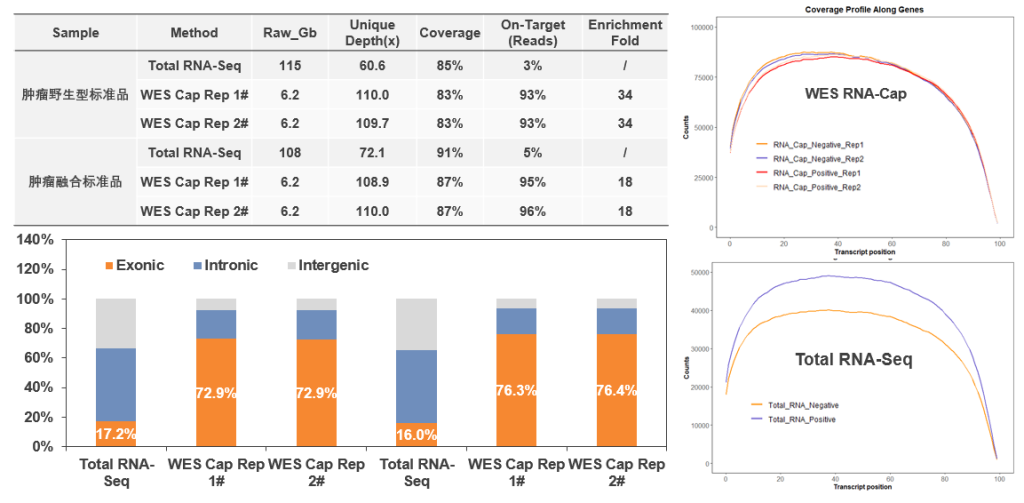
图4 Core Exome Panel v3.0 RNA-Cap与Total RNA-Seq基本表现比较
在融合基因检测性能上,WES Cap在6.2Gb测序数据量,平均深度为~110x的条件下,可以稳定地检出6种融合基因变异。
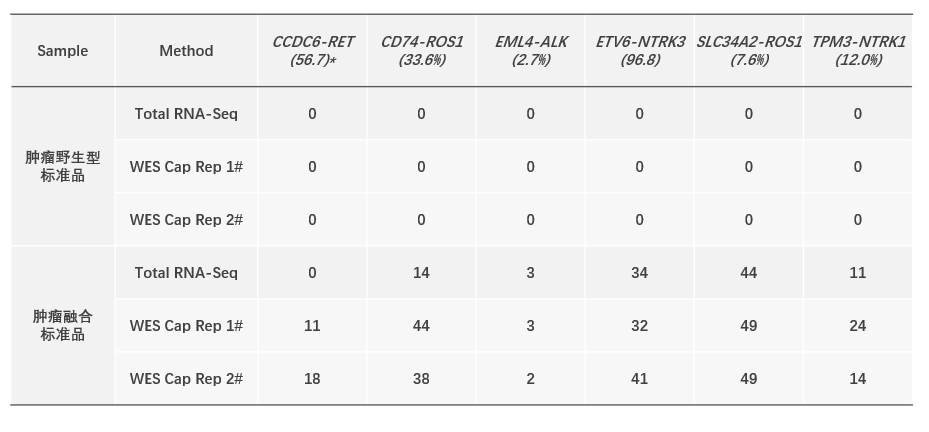
表1 融合基因检测情况;* 融合基因表达的qPCR检测结果(GW-OPSM001,LOT#202106001)




