上文提到,他汀类药物的降脂能力有限(LDL-C降低<50%),同时存在长期用药的依从性和一定的副作用等问题,因此,新型降脂药物的任务就是要解决这些问题。如下表所示,新型降脂药物-PCSK9抑制剂至少可以降低50%的LDL-C,这已经是他汀类药物的上限,而且还不用每天服药,依从性更好。
小编用亲身经历让大家感受一下这款新药的降脂效果,由于先前服用他汀类药物的降脂效果并不理想,随后开始使用他汀联合PCSK9抑制剂治疗,4个月后,小编的LDL-C从4.78 mmol/L降至1.32 mmol/L,降幅达到72%,可谓立竿见影。
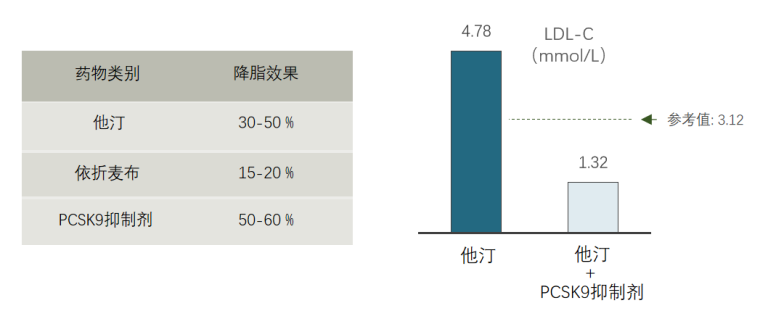
图1. 不同药物降脂效果[1]及他汀联合PCSK9抑制剂降脂案例
他汀药物和PCSK9抑制剂分别作用于胆固醇的合成和摄取过程,前者得益于胆固醇合成途径的研究,而PCSK9抑制剂的出现则是来自于科学家们与一种罕见遗传病-家族性高胆固醇血症(FH)的长期斗争,所谓知己知彼,才能克敌制胜,人类从FH这种自身疾病身上学习到了很多,并极大的促进了我们对人体胆固醇平衡机制的理解,而PCSK9抑制剂的出现则更像是基础研究的水到渠成。
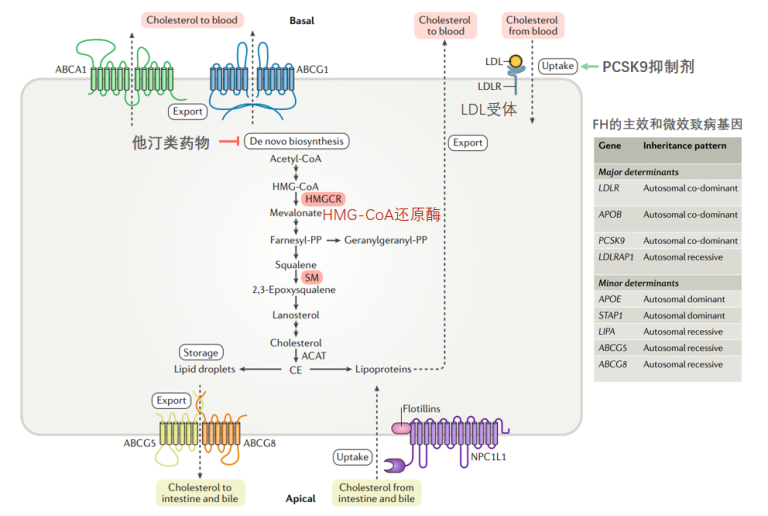
图2. 极化细胞中胆固醇代谢的主要通路以及他汀和PCSK9抑制剂的作用通路[2]
2023年1月,法国科学家Marianne Abifadel与Catherine Boileau(PCSK9基因发现的研究团队之一)在Journal Of Internal Medicine发表综述文章(《Genetic and molecular architecture of familial hypercholesterolemia》,该综述系统回顾了人类对FH近百年的研究历程,并梳理了FH相关致病基因的发现过程,以及分子遗传学研究对FH的诊断以及心血管疾病治疗的贡献[3]。
(1)1938年,Carl Muller首次报道遗传性高胆固醇血症及其相关症状;
(2)1964年,Khachadurian首次报道FH的遗传和表型研究;
(3)1974年,Goldstein和Brown发现LDL受体(LDLR),这也是第一个被发现的FH致病基因;
(4)1984年,Innerarity和Soria(1989年)等人发现第二个FH致病基因-载脂蛋白B(APOB);
(5)2003年,Abifadel等人发现第三个FH致病基因PCSK9。除了诊断之外,新基因的发现对治疗的影响也是决定性的。
(6)2015年,首个PCSK9抑制剂(单克隆抗体)获得批准,LDL-C显著降低到前所未有的水平(图3)。

图3. FH研究历程的关键节点[3]
家族性高胆固醇血症(FH)的特征包括低密度脂蛋白胆固醇(LDL-C)显著升高,特征性黄色瘤、早发心血管疾病家族史,可分为纯合子FH(HoFH)和杂合子FH(HeFH),HoFH的症状非常严重,在幼年时期就会发生心脏病并可能在30岁前去世,HoFH流行率为百万分之一,是名副其实的罕见病,有些医生一辈子都碰不到一例;杂合子FH(HeFH)的症状稍轻,但流行率却非常高,在中国人群中达到了1/200,因此,FH又并不“罕见”。

图4. 患有HoFH的10岁女孩,她在8岁时就发生了心肌梗死[4]
时间回到1973年,胆固醇代谢研究正在如火如荼的开展,人们认为胆固醇代谢主要发生在肝脏和肠道,同时还存在对HMG-CoA还原酶的反馈调节机制,想要研究人体的胆固醇代谢机制就必须获得这些细胞,但这显然不现实。为了解决这个问题,美国科学家戈尔茨坦(Joseph L Goldstein)和布朗(Michael S Brown)建立了一套更简便的体外细胞研究体系,利用容易获取的人体皮肤成纤维细胞,通过检测HMG-CoA还原酶的活性来间接了解胆固醇的合成。
他们很快发现,向细胞培养基中添加LDL能够抑制HMG-CoA还原酶活性,体系工作正常,但是添加HDL却不行,这暗示了一种LDL的特异性受体参与其中。与此同时,FH患者也找到了戈尔茨坦和布朗,希望能这项研究中找到治疗的希望。意想不到的是,HoFH患者的成纤维细胞对LDL完全不敏感(图5),似乎感受不到LDL的存在,强烈支持了LDL受体机制的存在。这一年远藤章团队刚刚分离出HMG-CoA还原酶抑制剂-美伐他汀。
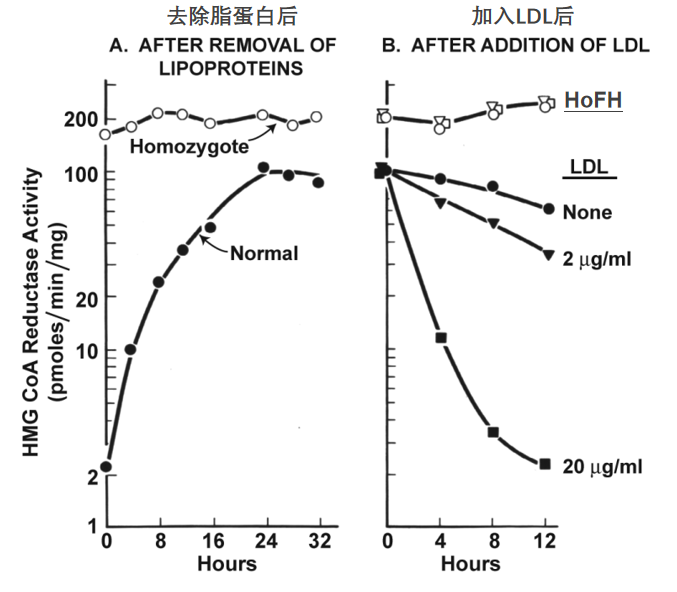
图5. HoFH患者的细胞对LDL没有反应[4]
1974年,同位素标记LDL实验表明,正常细胞表面存在LDL的结合位点,相反,HoFH患者的细胞则缺乏这些位点,该实验正式证明了LDL受体(LDLR)的存在。后续研究证明,细胞依靠LDLR识别LDL后通过胞吞作用从血液中摄取LDL(图6),随后,LDLR在胞内解离,并回到细胞表面;而大多数HoFH患者的细胞根本无法结合LDL,这就部分解释了FH患者具有高水平LDL-C的原因。
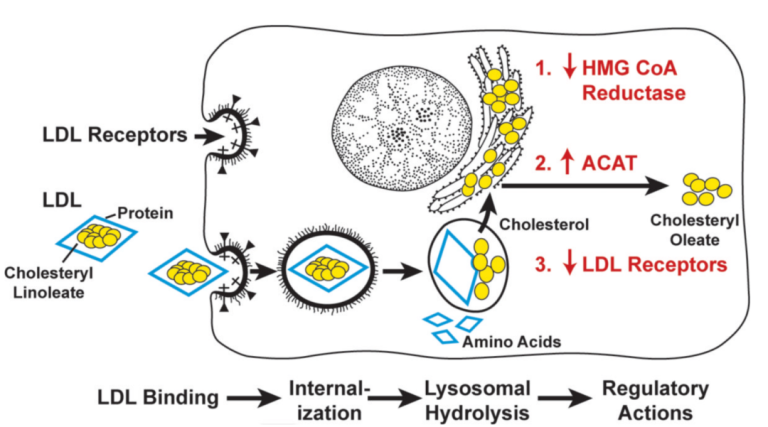
图6. 哺乳动物细胞低密度脂蛋白受体(LDLR)通路[4]
二十年后,膜结合转录因子SREBPs的发现进一步阐明了他汀类药物的作用机制(图7)-他汀类药物抑制胆固醇合成后,还会增强肝细胞对血浆中LDL的吸收。当摄入他汀类药物时,该药物主要进入肝脏,在那里它结合并抑制HMG-CoA还原酶,降低胆固醇的产生。胞内胆固醇水平的降低导致了SREBP片段进入细胞核,并激活LDLR和HMG-CoA还原酶的转录,从而增加了肝细胞膜上LDLR的数量。SREBPs也会同时增加HMG-CoA还原酶的表达量,但这并不会增加胆固醇的合成,因为该酶被他汀类药物抑制。新产生的LDLR从血液中去除LDL,并将其输送到细胞内部,在那里LDL被消化,其释放的胆固醇可用于代谢。净效果是肝脏中的胆固醇含量保持在正常水平,同时血液中的低密度脂蛋白胆固醇(LDL-C)水平保持在较低水平。幸运的是,LDLR不会与高密度脂蛋白(HDL)结合,因此这种有益的脂蛋白在血液的水平不会下降。
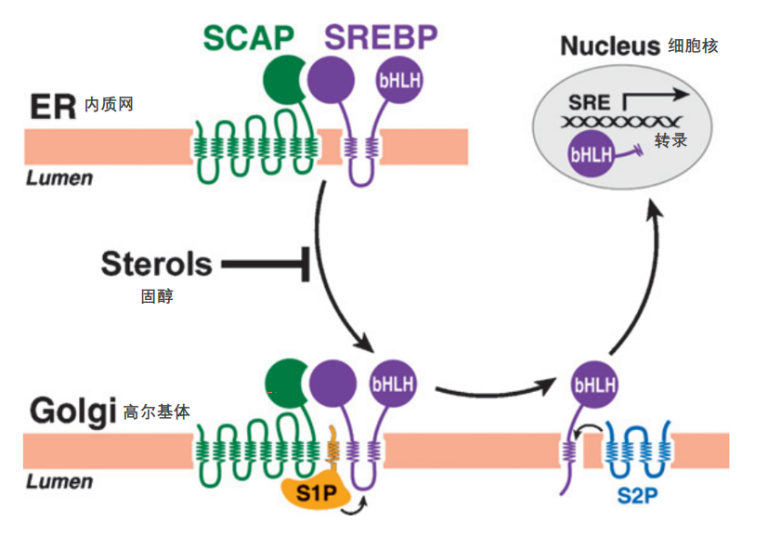
图7. SREBP通路[5]
戈尔茨坦和布朗戈尔茨坦的发现彻底改变了人们对胆固醇代谢的认识,他们的研究对于心血管疾病的诊治具有里程碑意义,1985年,戈尔茨坦和布朗因发现胆固醇代谢调节机理而荣获诺贝尔生理学或医学奖。

图8. 1985年10月15日,戈尔茨坦(左)和布朗(右)获得诺贝尔生理学或医学奖[4]
虽然发现了两个FH的致病基因(LDLR、APOB ),但仍然约有40%的FH患者致病基因不明确[1],一定还有其他基因导致了FH疾病。20多年前,法国科学家Catherine Boileau带领团队开始寻找FH其他致病基因。1999年,Boileau团队对法国FH家系的研究发现,人类1号染色体的短臂上(1p34.1-p32)可能隐藏着导致FH的“第三个致病基因”[6]。

图9. 第三个FH致病基因隐藏在人类1号染色体上[6、7]
但是两年过去了,由于当时的人类基因组图谱并不完整也没有高通量测序技术, Boileau团队在鉴定了几十个候选基因后,仍然没有找到答案。科学是需要灵感的(“猜”),就在他们一筹莫展的时候,另一个法国科学家Seidah发现了一个名为NACR1的基因,该基因在肝脏中具有高表达,并且正好位于染色体1p32区域内。
随后,两个团队研究证实,12.5%的FH患者家庭都存在NACR1基因变异(图10),它就是要找的第三个FH致病基因,并将NACR1正式命名为PCSK9(前蛋白转化酶枯草溶菌素9)[8],这一发现对于揭示PCSK9致病性变异在FH中的临床意义及其在胆固醇稳态调节中完全未知的作用至关重要[3]。
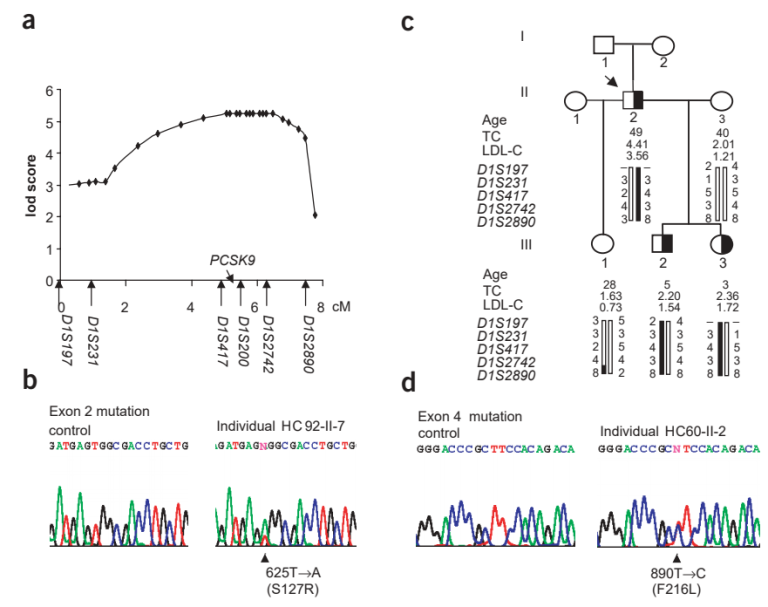
图10. 第三个FH致病基因-PCSK9被发现[8]
对于基因功能的研究,通常需要从正反两面进行证实,分别探索功能获得(GOF)和功能缺失(LOF)型基因变异对表型的影响。在小鼠中过表达PCSK9会导致LDL-C明显升高,表明Boileau团队发现的PCSK9变异可能属于GOF,后续研究证实他们发现的变异的确可以让PCSK9蛋白更加稳定。接下来,就该寻找LOF的证据了。
很快,Hobbs团队于2005年在美国非裔受试者中发现了PCSK9的两种无义突变(p.(Tyr142*)和p.(Cys679*)),2.6%的受试者携带了该变异,这些人较非携带者的LDL-C水平下降了40%,要知道,这种降脂效果已经超过常规的他汀疗效[8]。
紧接着,在2006年,更大规模的研究数据出炉,对随访近20年的动脉粥样硬化风险研究(ARIC)受试者进行基因测序和综合分析发现,在3363名黑人受试者中,有2.6%携带了PCSK9 LOF变异,其LDL-C水平和冠心病风险较非携带者分别降低28%和88%(图11);在9524名白人受试者,有3.2%携带了PCSK9 LOF变异,其LDL-C水平和冠心病风险较非携带者分别降低15%和47%。上述遗传学证据证明,PCSK9是调控血浆LDL-C水平的重要基因。
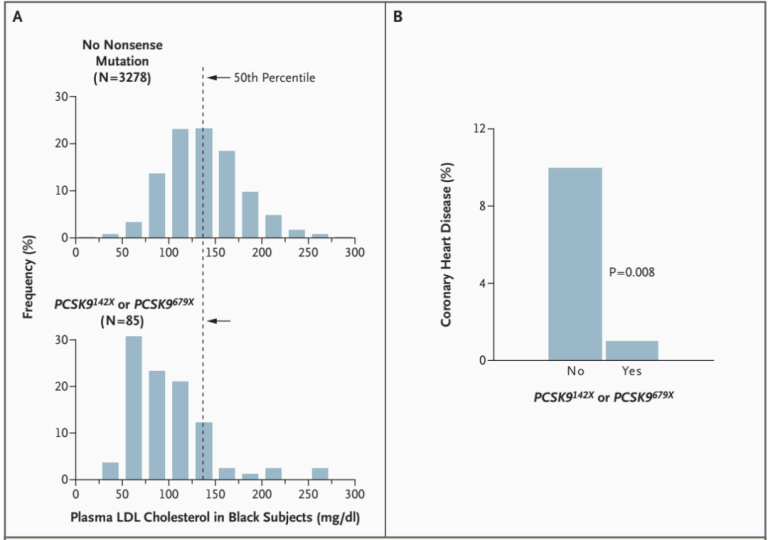
图11. PCSK9 LOF变异能够明显降低血浆LDL-C水平和冠心病风险[10]
在HMG-CoA还原酶被证明是胆固醇合成的限速酶后,寻找其抑制剂导致了他汀药物的发现。同样,PCSK9无疑是极具吸引力的降脂靶点。不过,上述Hobbs团队的功能缺失研究都是来自PCSK9的杂合突变,属于部分丧失功能,那么PCSK9完全消失会不会对人体造成其他负面影响就成了PCSK9药物开发的关键。
同年,Hobbs团队报道了一个携带了复合杂合型PCSK9变异的案例(II2),II2的血浆PCSK9蛋白完全检测不到,同时其LDL-C下降到了惊人的14 mg/dl,是正常水平的近1/10,更为重要的是II2非常健康,这个案例在一定程度上支持了靶向PCSK药物的安全性。这些证据充分说明,PCSK9可以调控LDL-C水平并影响心血管疾病风险,是降脂药物开发的潜在靶点。

图12. 携带复合杂合PCSK9变异的32岁美国非裔女性II2[11]
后续研究阐明了PCSK9的作用机制,LDL受体(LDLR)负责将血浆中的LDL运输到细胞内(图13 右),LDLR在其短暂的一生中(20个小时)会往返于细胞膜和胞质之间数百次[4],转运LDL以维持细胞内的胆固醇水平。PCSK9是一种分泌蛋白,与LDLR结合后可以将LDLR降解掉(图13 左),LDLR减少后,流入细胞的胆固醇自然就减少了,因此,PCSK9可以看作是胆固醇流入细胞的“闸门”。PCSK9失活后,LDLR降解被抑制,LDLR可以尽情的将血浆中的LDL转运进细胞,所以表现出血浆LDL-C水平明显降低。
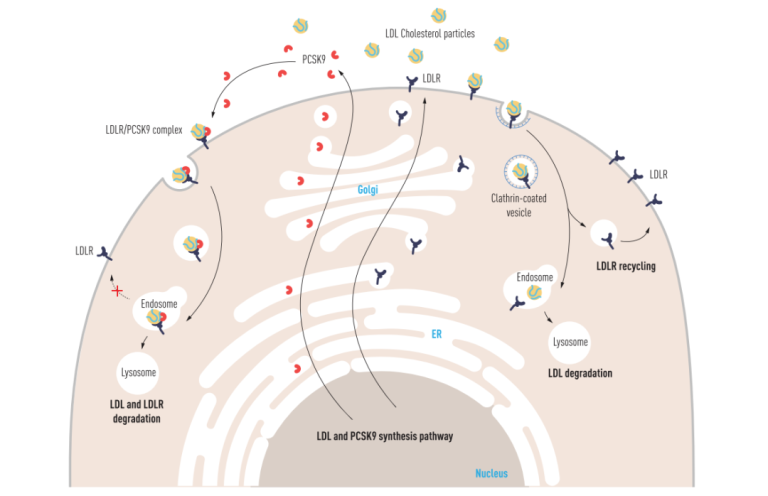
图13. PCSK9对LDLR的调控机制[3]
在PCSK9基因发现十二年后,第一个PCSK9抑制剂单抗药物于2015年上市,2021年底,FDA批准PCSK9 siRNA药物用于治疗动脉粥样硬化性心血管疾病(ASCVD)。在未来,口服抗PCSK9药物以及靶向其他基因的降脂疗法将进一步丰富我们对抗心血管疾病的武器库。
不论是他汀药物开发还是PCSK9抑制剂的出现,都离不开科学家们对胆固醇代谢机制孜孜不倦的探索;同时,FH疾病的分子遗传学研究对PCSK基因的发现发挥着举足轻重的作用,在PCSK9基因发现20周年之际,让我们向在此生命科学领域中辛勤耕耘的每一位工作者致以敬意。